
氮杂双环硝胺类含能材料的分子结构与性能的关系
2017-06-28 14:20周长肖
火炸药学报 2017年3期
周长肖,申 程,陆 明
(南京理工大学化工学院,江苏 南京 210094)
氮杂双环硝胺类含能材料的分子结构与性能的关系
周长肖,申 程,陆 明
(南京理工大学化工学院,江苏 南京 210094)
使用Gaussian03软件,采用密度泛函理论(DFT)在B3LPY/6-31++G (d,p)计算精度下,计算并研究了9个具有不同官能团以及母环结构的氮杂双环硝胺类含能化合物的分子结构,分析了其结构与性能的关系,优化了几何构型,通过计算得到了分子体积、能量等;以此为基础计算了该系列材料的密度、爆轰性能以及感度等数据。通过比较分子结构,从密度、生成热、爆轰性能以及撞击感度等方面,研究了不同官能团以及环系结构对该类含能材料性能的影响;通过对比得到了对双环硝胺化合物有利的官能团及结构。结果表明,双环硝胺结构有利于稳定结构以及形成紧凑的空间排布,并提高化合物的撞击感度及密度;同时,羰基的引入以及接近零氧平衡是提高该类含能材料爆炸性能的技术途径。
含能材料;双环硝胺;爆轰性能;DFT;密度泛函理论;高能量密度材料
引 言
随着现代战争的高速发展,武器系统对含能材料的性能提出了更高的要求,新型高能量密度材料(HEDM)的开发逐渐成为研究的重点[1-2]。目前,硝胺类化合物特别是环状硝胺,如 1,3,5-三硝基-1,3,5-三氮杂环己烷(RDX),1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷(HMX)等,对其分子结构进行修饰以提升性能的研究成为含能化合物合成的热点。研究人员将RDX、HMX作为模板或含能基底材料,进行了多种结构的改性和修饰,以提高其性能[3-6]。如Wang等[4-5]设计了1,3,4,5,7,8-六硝基八氢化二咪唑[4,5-b:4′,5′-e]吡嗪-2,6-(1H,3H)-N,N′-二亚硝胺 (ONIP);Shen等[7]设计了双环硝胺材料1,3,5,7,9,11-六硝基-1,3,5,7,9,11-六氮-螺[5.5]十一烷 (R4)等,理论密度大于1.9g/cm3,爆速大于9000m/s[8]。
然而,随着含能材料能量的不断提高,其感度也随之增大,迫切需要开发不敏感含能材料,提高武器的战场生存能力,以满足现代战争的需要。因此,在研究具有高能量密度、高分子稳定性的含能材料时,如果可以较为准确地预测其感度或相对稳定性,将会有效提高研究效率,最大程度地降低研究新型含能材料时的安全风险[9]。
本课题组在对K-56、ONIP[10]等项目的研究过程中发现,多环硝胺化合物的相对感度与分子结构有直接的关系,对于类似结构,母环的官能团、键长等结构差异对感度有直接影响,因此,本研究在已有的多环硝胺类含能材料的基础上,对9种双环或单环硝胺类含能材料的密度、爆轰性能以及撞击感度进行分析比较,归纳总结其中的规律,以期为设计潜在的高能低感HEDMs提供参考。
1 计算方法
1.1 研究对象
本研究对象为图1中的9种含能化合物,包括6种氮杂双环硝胺类含能化合物(化合物1~6)和构成这6种氮杂双环硝胺类含能化合物的3个单环结构化合物(化合物7~9)。6种氮杂双环硝胺类含能化合物分别是1,4,5,8-四硝基-1,4,5,8-四氮杂双环[4.4.0]癸烷(简称四硝基哌嗪,化合物1)、1,4,5,8-四硝基-1,4,5,8-四氮杂双环[4.4.0]癸烷、2,4,7,9-四硝基-2,4,7,9-四氮杂双环[4.3.0]壬烷(化合物2)、2,4,7,9-四硝基-2,4,7,9-四氮杂双环[4.3.0]壬酮-8(简称K-56,化合物3)、2,4,6,8-四硝基-2,4,6,8-四氮杂三环[3.3.0]辛烷(简称双环HMX,化合物4)、四硝基半甘脲(化合物5)和四硝基甘脲(化合物6)。以氮杂单环结构为基础设计氮杂双环或者多环结构,基本目的是增加环系以提升骨架刚性, 从而提高分子密度和能量水平[11]。
1.2 计算方法
量子化学计算均采用Gaussian03数据包[12],采用密度泛函理论(DFT)在B3LPY/6-31++G (d,p)计算精度下,对所设计的分子结构进行构型优化,并确保振动分析无虚频[13-15];体积计算使用Monte-Carlo统计方法,设定iop(6/45=2000)以减小统计方法因随机波动而导致的误差;在爆轰性能的计算中,使用等键方程计算化合物的生成热,其中母环结构均使用G2高精度方法计算,并结合Kamlet-Jacobs方程计算其性能数据[16]。所有计算收敛参数均采用程序内定值。
1.2.1 密度计算
运用分子表面静电势(ESP),采用公式(1)计算密度[19]:
(1)

1.2.2 生成热计算
采用设计等键反应法[7]计算生成热,设计的反应如下:
R(NO2)n+nCH4→RHn+nCH3NO2
基于上述方程,生成热可由式(2)计算得到:
ΔH298=∑ΔHf(g, P)-∑ΔHf(g, R)=
∑ΔHf(P)-∑ΔHf(R)
(2)式中:∑ΔHf(P)为产物在298K下的气相生成热,∑ΔHf(R)为反应物在298K下的气相生成热,CH3NO2以及CH4的生成热由文献获得[18],母环RHn的气相生成热的计算均利用原子化反应法并使用G2高精度算法计算得到,以保证精度[7]。
通常含能材料都是室温下的固体化合物,因此具有实际意义的是固相生成热,根据盖斯定律可知[19]:
ΔHf(solid)= ΔHf(gas)-ΔH(sub)
(3)
式中:ΔH(sub)为升华焓,可以使用Politzer等[20]提出的式(4)进行计算:
(4)
1.2.3 爆轰性能计算
爆速、爆压及爆热采用Kamlet-Jacobs公式[16]预测:
(5)
(6)

对于上述9种CaHbOcNd类型的化合物,均满足b/2≤c≤(2a+b)/2,此时有:
N= (b+2c+2d)/4M
(7)
(8)
Q×103=(28.9b+94.05(c/2-b/4)+
(9)

1.2.4 撞击感度计算
采用Miroslav等[22]报道的方法预估撞击感度H50(2.5 kg),其计算公式如下:
2结果与讨论
2.1 分子结构与密度的关系
采用公式(1)计算9种含能化合物的密度,结果见表1。
由表1可以看出,双环化合物1~6的密度均大于单环化合物7~9,说明双环有利于结构的紧凑。化合物3、5和6的密度均大于1.9g/cm3,是潜在的高能量密度材料。通常认为,硝基的存在是提高化合物密度的关键因素,然而,化合物1~6均含有4个硝基,密度范围为1.76~1.94g/cm3,波动幅度很大。 因此,除硝基数量外,应还有其他因素与密度有紧密关联。
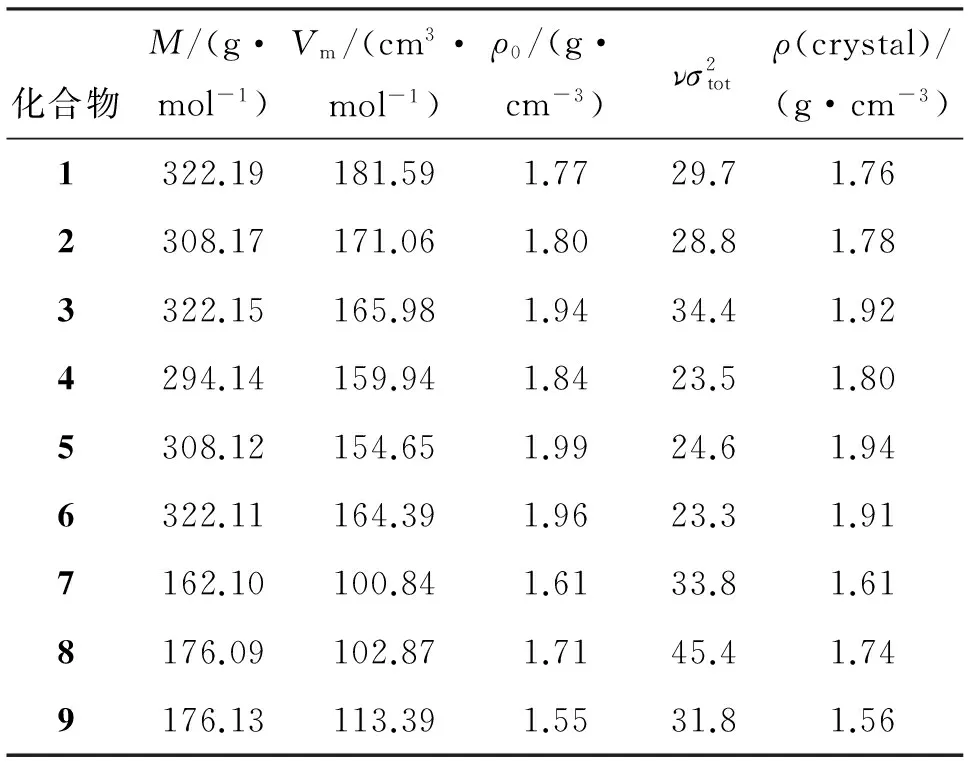
表1 9种氮杂环含能化合物的理论密度Table 1 Theoretical densities of 9 kinds of aza-cyclic energetic compounds
在结构相似的双环化合物中,母环结构的紧凑与否很大程度上决定了密度的高低。例如,六元环并六元环结构(化合物1)由于其六元环的结构比较松散,环平面容易扭曲,空间结构不紧凑,导致密度较低,这种观点在具有相似结构的单环化合物7和9上也能很好地得到验证,饱和五元环化合物(化合物7)的密度大于六元环化合物(化全物9)。
在上述基础上进一步比较官能团对密度的影响。由表1可知,在具有相同类型的母环结构的3个系列化合物内部比较可以发现,亚甲基越多密度越低,羰基可以提高密度[23],这是由于亚甲基增加了环结构翻转的可能,导致整体结构松散,而含不饱和键的羰基则可以使环的刚性增加从而更紧凑。因此,含3个亚甲基的五并六元环结构化合物(化合物2)的密度低于其他五并六元环结构。
对设计的9种化合物在Gaussian03软件平台下进行几何构型优化,计算出能量最低的构型。考虑到所有计算数据均在几何构型优化的基础上完成,选用计算精度较好且计算效率也较高的密度泛函理论(DFT)下的B3LYP算法进行计算,所得分子几何构型如图2所示。
在此基础上,得到相应分子最稳定构型下的结构数据,母环的平均键长数据如表2所示。

化合物键长/nmC-CC-N平均10.1534650.1468590.14866020.1541670.1466500.14815430.1545910.1456200.14741440.1568440.1464240.14758250.1573760.1452070.14655960.1557920.1440220.14533070.1530020.1469960.14819780.1536120.1455980.14740190.1538720.1471080.149363
由表2可以看出,化合物3、5、6的平均键长在双环结构中是最短的,说明化合物3、5、6的环结构更紧凑,具有更大的密度,同时化合物3、5和6的分子均含有羰基,化合物5和6均为五并五元环结构,与上文的论述较为一致。此外,化合物8虽然平均键长也很短,但是化合物8是单环结构,与双环结构相比不利于分子体积的压缩,导致密度偏低,可见并环结构对密度提升也有很大的增益。同时由表1也可以看到,具有五元环以及羰基结构的化合物8在单环化合物7、8、9中也具有较高的密度,进一步佐证了本文的观点。
2.2 结构与生成热的关系
9种化合物的总能量(E0)、零点能(ZPE)、热修正值(ΔHT)和生成热(ΔHf)计算数据见表3。
由于表3得到的是气相生产热,而更具实际意义的是固相生成热,可由文献[20-21]的公式计算得到升华焓与固相生成焓,结果见表4。

表3 9种含能化合物的总能量、零点能、热修正值和 生成热的计算Table 3 Total energies (E0), zero-point energies (ZPE), thermal corrections (ΔHT),heat of formation of 9 kinds of energetic compounds and HOFs
注:(1)ZPE 的计算校正因子为0.98;(2)ΔHT的计算校正因子为0.96[24]。

表4 9种含能化合物的升华焓与固相生成焓的计算Table 4 Heat of sublimation and solid phase HOF of 9 kinds of energetic compounds

从表4数据可以看出,单环结构的能量普遍比双环低,双环具备更高的生成热,这与双环结构更大的环张力密不可分,进一步说明,双环结构有利于结构的压缩,提高密度及生成热。同时,具有羰基的化合物3和5的生成热明显比同样双环结构的其他分子低,说明羰基化合物一定程度上降低了结构的能量,这可能对提高分子稳定性有一定的帮助。
结合校正之后的固态生成热以及晶体密度,可以进一步分析相应结构的爆轰性能。
2.3 结构与爆轰性能的关系
使用Kamlet-Jacobs公式[16]计算得到化合物的爆轰性能,结果见表5。

表5 9种含能化合物的爆热、爆速和爆压的计算值Table 5 Heat of detonation (Q),detonation velocities (D), and detonation pressures (p) of 9 kinds of energetic compounds
从表5可以看到,9种化合物的爆速均超过7600m/s。结合密度(表1)与固体生成热(表4)的结果可以看到,化合物3、5和6的密度最高,爆速超过9000 m/s,因此其爆轰性能最好。
此外,通过比较氧平衡可以看出,化合物5和6的氧平衡数值最接近零氧平衡,好的氧平衡可以提高炸药的爆热,也就保证了化合物的能量输出。虽然与化合物3、5、6相比,化合物4和6的密度不高,但其爆轰性能却很好,说明氧平衡也是衡量含能材料性能的重要指标。
2.4 结构与感度性能的关系
图3为化合物3(五并六元环含羰基)、6(五并五元环含双羰基)、8(五元单环含羰基)的静电势分布图。图中红色数值代表静电势的极小值点,黑色数值代表静电势的极大值点。
从图3中可以看出,具有类似双环结构的化合物3和6静电势变化幅度明显,化合物6(-86.69~281.23kJ/mol)大于化合物3(-111.26~235.41kJ/mol),而对于单环结构而言虽然静电势变化幅度不大,但是由于分子结构小,静电势的表面积小,静电势在分子表面变化幅度仍然较大,分子稳定性与双环结构相比没有优势。同时,从图3可以看出,羰基结构无论在单环还是双环上都是静电势的极小点,从静电势的角度分析,对结构稳定性不利。
9种化合物的理论感度计算结果见表6。

表6 9种含能化合物撞击感度的计算结果Table 6 The calculation results of impact sensitivities for 9 kinds of energetic compounds
从表6看出,化合物3、5、6和8均有羰基结构,感度相对较高,但均优于RDX(H50为26cm)以及HMX(H50为29cm)[10,25]等传统的炸药,化合物1~6不但具有紧凑的结构,同时还有较低的环张力,具有十分优良的撞击感度,是潜在的钝感含能材料。同时结合爆轰性能数据,表5中化合物3、5、6具有和RDX相当的爆轰性能,是潜在的钝感含能化合物。
3 结 论
(1)对6个双环硝胺类含能化合物及3个单环结构进行了量子化学计算。通过对比,6个双环硝胺含能化合物具有良好的爆轰性能及优异的感度,其中化合物3、5与6撞击感度分别为108、75、74cm,密度超过1.9g/cm3,爆速大于9000m/s,爆轰性能优于RDX,与HMX相当,是钝感高能密度材料。
(2)通过对比密度、生成热、爆轰性能以及感度与结构的关系,紧凑的双环结构有利于提升化合物的密度以及能量水平;羰基的存在可以进一步提高环结构的紧凑程度,但同时会降低生成热;亚甲基会极大地降低密度,设计分子时应当避免。
(3)氮杂双环结构的稳定性较好,在钝感含能化合物中具有良好的研究价值。
[1] 李亚南,毕福强,王伯周,等.基于1,5-二硝胺基四唑高能材料的合成及性能[J].火炸药学报,2015,38(6):16-20. LI Ya-nan,BI Fu-qiang,WANG Bo-zhou,et al.Synthesis and properties of high energy materials based on 1,5-di(nitramino)tetrazole[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2015,38(6):16-20.
[2] 邵航松,凌亦飞,侯天骄,等.9-硝基-9-氮杂双环[3.3.1]壬烷-2,6-二醇二硝酸酯的合成与表征[J].火炸药学报,2016,39(2):64-67. SHAO Hang-song,LING Yi-fei,HOU Tian-jiao,et al.Synthesis and characterization of 9-nitro-9-azabicyclo[3.3.1]nonane -2,6-diyl dinitrate[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2016,39(2):64-67.
[3] Feng W, Xu X, YANG Ya, et al. Research process on preparation and application in propellant of graphene[J].Journal of Ordnance Equipment Engineering, 2016(4):89-94.
[4] Yang J, Wang J, Gao Z, et al. Research on charge diameter′s influence to SCO character of HMX base explosives[J]. Journal of Sichuan Ordnance, 2015(6):117-119.
[5] Xu Y, Shen C, Lin Q, Wang C, et al. 1-Nitro-2-trinitromethyl substituted imidazoles: a new family of high performance energetic materials[J]. Journal of Materials Chemistry A, 2016, 4(45):17791-17800.
[6] Davin G P, David E C, Brian L S, et al, An energetic triazolo-1,2,4-triazine and its n-oxide[J]. Angewandte Chemie International Edition, 2016, 55:15315-15318.
[7] Shen C, Wang P C, Lu M.Molecular design and property prediction for a series of novel dicyclic cyclotrimethylene trinitramine(RDX)-derivatized as high energy density materials [J]. Journal of Physical Chemistry A, 2015, 119 (29): 8250-8255.
[8] Duan W D , Huang F L. Investigation on the exploding security performance of low vulnerability explosive nitroguanidine [J].Journal of Projectiles, Rockets, Missiles and Guidance,2002, 23(4): 167-171.
[9] Frisch M J, Tracks G W, Schlegel H B, et al.Gaussian 03, Revision C.02. [M].Wallingford CT: Wallingford Gaussian Inc, 2004.
[10] 申程, 王鹏程, 赵国政,等.1,3,4,5,7,8-六硝基八氢化二咪唑[4,5-b:4′,5′-e] 吡嗪-2,6-(1H,3H)-N,N′-二亚硝胺反应中间体的量子化学研究[J]. 高等学校化学学报, 2015, 36(1): 142-148. SHEN Cheng, WANG Peng-cheng, ZHANO Guo-zheng,et al. Computational study on the derivatives of N,N′-(1,3,4,5,7,8-hexanitrooctahydro-diimidazo [4,5-b:4′,5′-e]pyrazine-2,6(1H,3H)-diylidene)dinitramide[J].Chemical Journal of Chinese Universities, 2015, 36(1): 142-148.
[11] Qiu L, Gong X D, Xiao H M. Theoretical studies on thermolysis mechanism and stability of trans-1,4,5,8- etranitro- 1,4,5,8-tetraazadecalin isomers[J]. Chinese Journal of Chemistry, 2008, 26: 2165-2172.
[12] 舒远杰. 高能硝胺炸药的热分解[M]. 北京:国防工业出版社, 2010.
[13] Zhang Qinghua, Zhang Jiaheng, Qi Xiujuan .Molecular design and property prediction of high density polynitro[3.3.3]-propellane-derivatized frameworks as potential high explosives [J]. Journal of Physical Chemistry A, 2014, 118: 10857-10865.
[14] 杨尧,申程,陆明.1′-二羟基-5,5′-联四唑-5-氨基四唑盐的合成及性能预估[J].火炸药学报,2014,37(5):9-13. YANG Yao,SHEN Cheng,LU Ming.Synthesis and property prediction of 5-aminotetrazolium 1-hydroxy-1H,1′-H-5,5′-bitetrazol-1-olate[J].Chinese Journal of Explosives & Propellants(Huozhayao Xuebao),2014,37(5):9-13.
[15] 毕福强,翟连杰,张俊林,等.2,4,6,8-四硝基-2,4,6,8-四氮杂三环[3.3.0.03,7]辛烷的理论研究[J].火炸药学报,2016,39(6):26-31. BI Fu-qiang,ZHAI Lian-jie,ZHANG Jun-lin,et al.Theoretical study of cage-HMX: 2,4,6,8-tetranitro-2,4,6,8-tetraazatricycle [3.3.0.03,7]octane[J]. Chinese Journal of Explosives & Propellants( Huozhayao Xuebao), 2016,39(6):26-31.
[16] Kamlet M J, Jacobs S T. Chemistry of detonation. I. A simple method for calculating detonation properties of C, H, N, O explosives [J]. Journal of Chemicial Physics,1968, 48(1):23-25.
[17] Politzer P, Martinez J, Murray J S,et al. An electrostatic interaction correction for improved crystal density prediction [J]. Molecular Physics, 2009, 107(19):2095-2101.
[18] Nicolaides A, Rauk A, Glukhovtsev M N, et al.Heats of formation from G2,G2(MP2) and G2(MP2,SVP) total energies [J].Journal of Physical Chemistry,1996,100:17460-17464.
[19] Atkins P W. Physical Chemistry [M]. Oxford:Oxford University Press,1982.
[20] Politzer P, Murray J S, Grice M E M, et al, Calculation of heats of sublimation and solid phase heats of formation [J]. Molecular Physics, 1997, 91: 923-928.
[21] Politzer P, Murray J S. Some perspectives on estimating detonation properties of C, H, N, O compounds [J]. Central European Journal of Energetic Materials, 2011, 8: 209-220.
[22] Pospíšil M, Vávra P, Concha M C, et al, A possible crystal volume factor in the impact sensitivities of some energetic compounds[J].Journal of Molecular Modeling, 2010, 16: 895-901.
[23] Zhang Junlin, Xiao Chuan, Zhai Lianjie, et al, Synthesis and properties of the fused aza-polynitrocyclic compounds[J]. Chinese Journal of Organic Chemistry, 2016, 36: 1197-1207.
[24] Scott A P, Radom L. Harmonic vibrational frequencies: an evaluation of Hartree-fock, mφller-plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors[J].Journal of Physical Chemistry, 1996, 100: 16502-16513.
[25] Yin P, Mitchell L A, Parrish D A, et al.Energetic n-nitramino/n-oxyl-functionalized pyrazoles with versatile p-p stacking: structure-property relationships of high-performance energetic materials[J].Angewandte Chemie International Edition, 2016, 55: 14409-14411.
Relationship between Molecular Structure and Properties of the Energetic Materials of Aza-bicyclic Nitramine
ZHOU Chang-xiao, SHEN Cheng, LU Ming
(School of Chemical Engineering,Nanjing University of Science and Technology, Nanjing 210094, China)
The molecular structures of energetic materials of nine aza-bicyclic nitramine with different functional groups and parent ring structure were calculated and studied under B3LYP 6-31++G (d, p) calculation accuracy using density fuctional theory (DFT) with Gassian03 software. The relationship between the structure and properties was analyzed. Their geometry configurations were optimized. The molecular volume and energy etc. were obtained via. calculation. Based on this, the density, detonation properties and sensitivities of the materials were calculated. Through comparison of the molecular structure, the effects of different functional groups and structure of rings on the properties of this kind of energetic materials were studied from the aspects of density, heat of formation, detonation properties and stability etc. The functional groups and structure of bicyclic nitramine compounds were obtained via. comparison. The results show that bicyclic nitramine structure is conducive for stable structure, formation of a compact space layout and improvement of impact sensitivity and density. At the same time, the introduction of carbonyl group and approaching zero oxygen balance are good technology process to improve the performance of such kind of energetic materials.
energetic materials; bicyclec nitramine; detonation property; DFT;density functional theory;high energy density material
10.14077/j.issn.1007-7812.2017.03.003
2017-01-15;
2017-03-14
国家自然科学基金委员会和中国工程物理研究院联合基金资助项目(No.U1530101)
周长肖(1989-),男,硕士研究生,从事含能材料合成研究。E-mail:502235882@qq.com
陆明(1963-),男,教授,从事含能材料的设计与合成研究。E-mail:luming@mail.njust.edu.cn
TJ55;O621
A
1007-7812(2017)03-0021-06
猜你喜欢
科学导报(2022年41期)2022-07-13
安徽农学通报(2022年8期)2022-05-06
天津建设科技(2022年2期)2022-05-05
动物营养学报(2022年1期)2022-02-20
科技信息·学术版(2021年20期)2021-11-02
火炸药学报(2020年5期)2020-10-27
云南医药(2020年5期)2020-10-27
固体火箭技术(2019年4期)2019-09-10
火工品(2019年3期)2019-09-02
火炸药学报(2018年4期)2018-09-01
