
原发性浆细胞白血病与淋巴瘤伴浆细胞增多的诊断与鉴别诊断
2017-06-24 14:12:10张会超王芃堉陈砚凝丁雅雯高社军
临床与实验病理学杂志 2017年5期
张会超,黄 晨,王芃堉,李 宏,陈砚凝,张 红,丁雅雯,高社军
原发性浆细胞白血病与淋巴瘤伴浆细胞增多的诊断与鉴别诊断
张会超,黄 晨,王芃堉,李 宏,陈砚凝,张 红,丁雅雯,高社军
目的 探讨原发性浆细胞白血病(primary plasma cell leukemia, PPCL)及淋巴瘤伴浆细胞增多的临床病理特征、诊断及鉴别诊断。方法 采用临床资料及细胞形态学、流式细胞术、免疫固定电泳及免疫组化EliVision两步法等检测7例PPCL及3例淋巴瘤伴浆细胞增多,并进行分析。结果 7例PPCL及3例淋巴瘤伴浆细胞增多的临床特点均以进行性贫血、血小板减少、发热、肝脾及淋巴结肿大最为常见;外周血细胞形态学分类浆细胞比例均大于20%,且伴形态学异常;外周血流式细胞免疫表型显示7例PPCL均表达CD38及CD138,2例表达CD56,2例表达CD20,轻链(Lamda、Kappa)均呈单克隆限制性表达,符合PPCL诊断;3例淋巴瘤伴浆细胞增多CD19、CD45呈弱阳性,CD38、CD138呈阳性,轻链IgL未见限制性表达,属于正常浆细胞的免疫表型。3例轻链(Ig)未见限制性表达,经淋巴结切除活检病理学检查确诊血管免疫母细胞性T细胞淋巴瘤2例,CD30阳性窦内大B细胞淋巴瘤1例。结论 PPCL与淋巴瘤伴浆细胞增多有相同的临床表现及相似的细胞形态学特征,PPCL的诊断需结合免疫固定电泳及流式细胞免疫表型;而淋巴瘤伴浆细胞增多还需结合淋巴结组织学检查才能确诊。
原发性浆细胞白血病;淋巴瘤;反应性浆细胞增多症;免疫组织化学
浆细胞白血病(plasma cell leukemia, PCL)是由浆细胞恶性增生引起的疾病,属于罕见的白血病类型,按形态学诊断标准当外周血中浆细胞数大于2×109/L或浆细胞比例大于20%并伴形态学异常时即可诊断[1]。WHO(2008)造血与淋巴组织肿瘤分类将原发性浆细胞白血病(primary plasma cell leukemia, PPCL)定义为单克隆的浆细胞[2]。PPCL可发生于多发性骨髓瘤的晚期,即继发性浆细胞白血病(secondary plasma cell leukemia, SPCL),也可在发病前无任何浆细胞疾病的征象,此种即PPCL。本文回顾性分析7例PPCL及3例淋巴瘤伴浆细胞增多的临床病理特征,以提高对该病的认识。
1 材料与方法
1.1 临床资料 收集2008年4月~2016年1月河北医科大学第四医院入院治疗的7例PPCL及3例淋巴瘤伴浆细胞增多。所有病例外周血中浆细胞比例均大于20%,且伴形态异常,既往无多发性骨髓瘤或其它浆细胞疾患史。其中男性6例,女性4例,年龄最大者67岁,最小者35岁,中位年龄57岁。首发症状以肿大淋巴结为主,其次为乏力、发热、腰痛,有骨质破坏者4例,淋巴结肿大8例,脾大者8例,肝大者4例,3例Coombs试验阳性(表1)。8例白细胞增高,10例均有不同程度贫血,8例有血小板减低;10例均有不同程度的免疫球蛋白增高,7例β2MG增高,6例LDH增高,6例Bun增高,7例CREA增高(表2)。
1.2 方法
1.2.1 骨髓细胞形态学检查 患者髂后上棘行骨髓穿刺,穿刺过程中避免骨髓穿刺液被血液稀释,选2块小粒较多、厚薄均匀的骨髓片,经瑞氏染色,然后油镜观察骨髓造血细胞及其它细胞形态。
1.2.2 流式细胞学检查 流式细胞仪为Beckman Coulter公司的Epics XL型,488 nm激发光源。单克隆抗体试剂购自美国Beckman Coulter公司。采用Expo32-ADC软件分析,设阴性对照,结果判断以细胞抗原表达>20%为阳性。取外周血细胞经淋巴细胞分层液处理后,调整细胞数为106/mL,置于PBS液中待用,单细胞悬液中加入单抗:CD45、CD19、CD56、CD30、CD38、CD138、CD23、CD20、FMC7、CD79b、CD5、cKappa、cLamda、cCD79a、CD3。4 ℃暗处孵育,用PBS洗浴3次,24 h内待检。

表1 患者的临床资料
CD30+SLBCL:CD30阳性窦内弥漫大B细胞淋巴瘤;ATCL:血管免疫母细胞性T细胞淋巴瘤
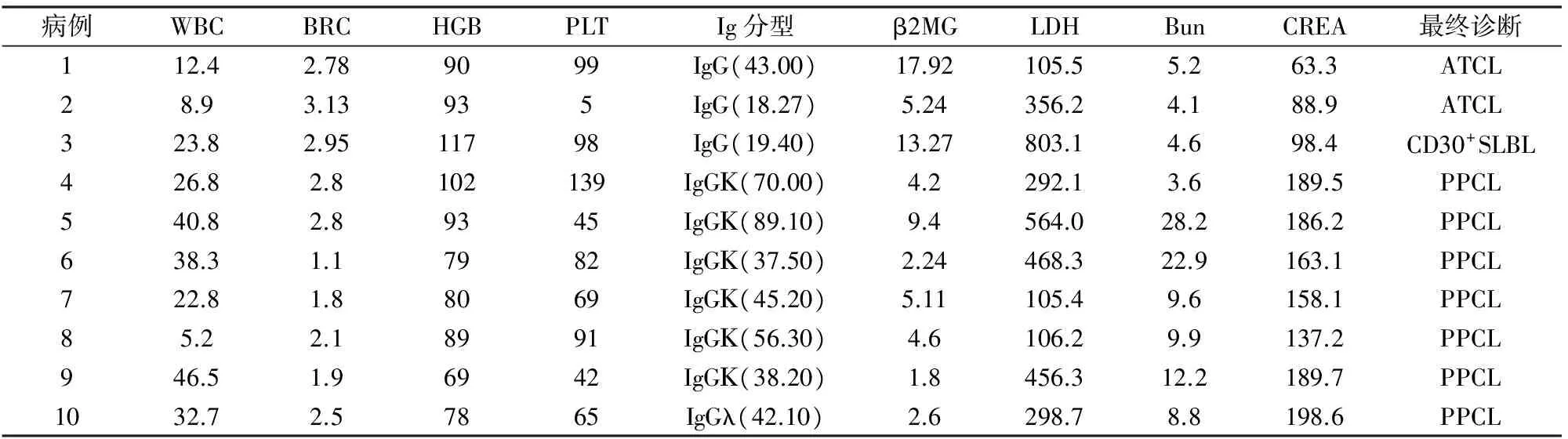
表2 患者的血常规、生化及免疫球蛋白数据
CD30+SLBCL:CD30阳性窦内弥漫大B细胞淋巴瘤;ATCL:血管免疫母细胞性T细胞淋巴瘤
1.2.3 免疫固定电泳法 法国Sebia HYDRASYS全自动电泳仪及原装试剂检测血清蛋白电泳及免疫固定电泳。血清蛋白电泳:取10例血清标本于加样梳在琼脂糖凝胶板上电泳、干燥、染色、脱色、烘干、扫描。免疫固定电泳:血清标本的M蛋白浓度预稀释成1.5 g/L再进行免疫固定电泳,取已稀释好的血清标本10 μL于加样梳上,在琼脂糖凝胶板上电泳后,加入抗人全血清40 μL,依次加入抗IgG、抗IgA、抗IgM、抗κ轻链,抗λ轻链血清各25 μL。孵育固定沉淀蛋白,再吸去多余的抗血清、烘干、染色,脱色、烘干,观察结果。
1.2.4 免疫组化法 标本采用10%中性福尔马林固定,石蜡包埋,4 μm厚切片,HE染色,光镜观察。免疫组化采用EliVision两步法染色,高压煮沸抗原修复,CD2、CD3、CD4、CD10、CD20、CXCL-13、CD5、Ki-67、CD30、CD7、CD15、Pax-5、BCL-6、PD-1、CD21、MUM-1及二抗、三抗,均购自福州迈新公司。DAB染色,HE复染,EBER原位杂交。
2 结果
2.1 细胞形态学 10例患者骨髓涂片:原、幼浆细胞百分比的中位值为67%(18%~88%);血涂片:原始或幼稚浆细胞明显增多,百分比的中位值为61.5%(15%~96%)(表3)。例4~例10骨髓及外周血原幼浆细胞比例明显增高,而例1~例3原、幼浆细胞比例较低。例4~例10浆细胞形态细胞大小不等,核呈圆形或椭圆形居中或偏位,染色质呈粗颗粒状,核仁可见1~2个,胞质丰富,内有灰蓝色细小颗粒及大小不等空泡;例1~例3的浆细胞形态呈圆形或椭圆形大小不等,个别有双核、巨大核,核仁模糊不清,核染色质较致密,核居中或偏位,胞质量中等,染色质染成深蓝色。
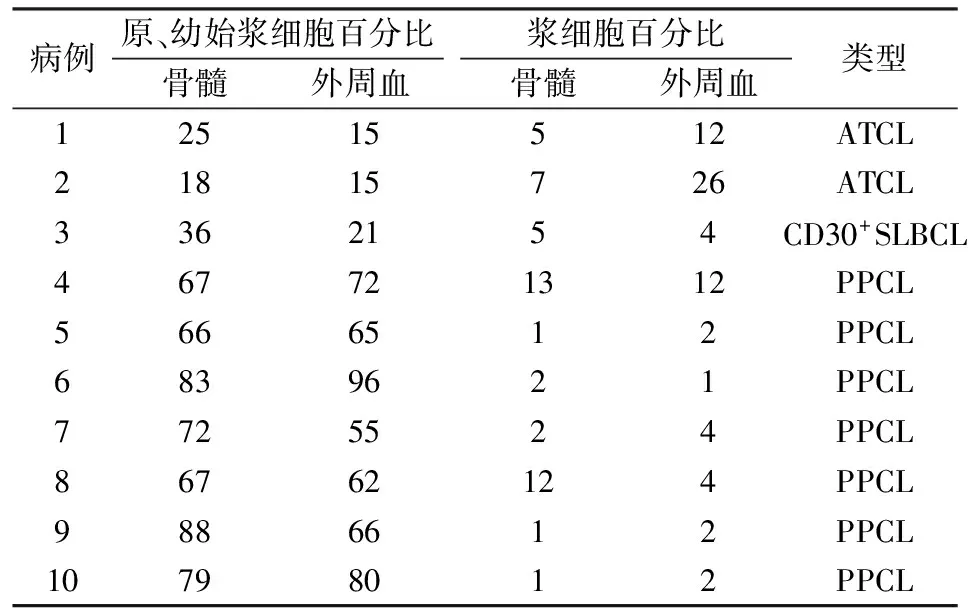
表3 患者骨髓及外周血中原、幼浆细胞比例(%)
CD30+SLBCL:CD30阳性窦内弥漫大B细胞淋巴瘤;ATCL:血管免疫母细胞性T细胞淋巴瘤
2.2 流式细胞术检测 例4~例10浆细胞的免疫组化标记均表达CD38、CD138,2例表达CD56,2例表达CD20,轻链(Lamda、Kappa)均呈单克隆限制性表达,6例呈Kappa限制性表达,1例呈Lamda限制性表达,符合恶性浆细胞的免疫表型。例1~例3浆细胞免疫表型:CD19、CD45均呈弱表达,CD38、CD138表达,轻链Lamda及Kappa均呈阳性,属于正常浆细胞的免疫表型。
2.3 免疫固定电泳 例4~例10血清免疫固定电泳显示存在单克隆免疫球蛋白;其中6例IgG(IgGΚ 5例、IgGλ 1例),1例IgAΚ;其中例1~例3均未见单克隆球蛋白。
2.4 淋巴结组织学特征 对3例血清免疫固定电泳呈多克隆者行淋巴结切除活检进行病理学检查。例1、2组织病理学特征:淋巴结正常结构被破坏,可见分支状的高内皮小静脉,副皮质区可见多形性的异型淋巴细胞浸润灶,该细胞小至中等大小,胞质淡染或透明(图1),包膜清晰,细胞异型较轻微,位于被破坏的滤泡旁或高内皮小静脉旁,混杂有数量不等的反应性小淋巴细胞、嗜酸性粒细胞、浆细胞及组织细胞。例3组织形态学特征:淋巴结病变均以淋巴窦明显扩张、肿瘤细胞选择性浸润并充斥窦腔为主要特征,扩张的淋巴窦呈圆巢形、裂隙状或不规则的分支吻合状(图2);瘤细胞呈多边形或圆形,彼此之间镶嵌排列,黏附性生长,细胞质丰富,嫌色或弱嗜酸性,胞质境界清楚或略显不清,细胞核大,圆形、椭圆形或略呈多形性,核膜清晰,染色质粉尘状或凝集成细颗粒状,有1~2个位置居中、显著嗜酸性大核仁呈典型的免疫母细胞形态,并伴大量成熟浆细胞及上皮样小静脉或薄壁小血管增生,残存少量滤泡结构,但明显萎缩。2.5 免疫表型 例1、2免疫表型:CD3(图3)、CD4、CD10、BCL-6、CXCL-13、PD-1均呈阳性,CD21(图4)均显示紊乱的滤泡树突状细胞网,Ki-67增殖指数均约40%;例1的EBER原位杂交阳性:15~20/40 HPF,例2的EBER原位杂交阳性:10~15/40 HPF;例1、2诊断为血管免疫母细胞性T细胞淋巴瘤。例3免疫表型:淋巴窦内肿瘤性细胞CD30(图5)、CD20(图6)、Pax-5及MUM-1均呈弥漫阳性,Ki-67增殖指数大于80%;该例诊断为CD30阳性的窦内弥漫大B细胞淋巴瘤。
2.6 最终诊断 结合骨髓细胞形态学、外周血流式细胞免疫表型最终诊断7例PPCL;结合淋巴结组织学、骨髓细胞形态学、外周血流式细胞免疫表型及免疫固定电泳最终诊断2例血管免疫母细胞性T细胞淋巴瘤伴浆细胞增多症,1例CD30阳性的窦内弥漫大B细胞淋巴瘤伴反应性浆细胞增多症。
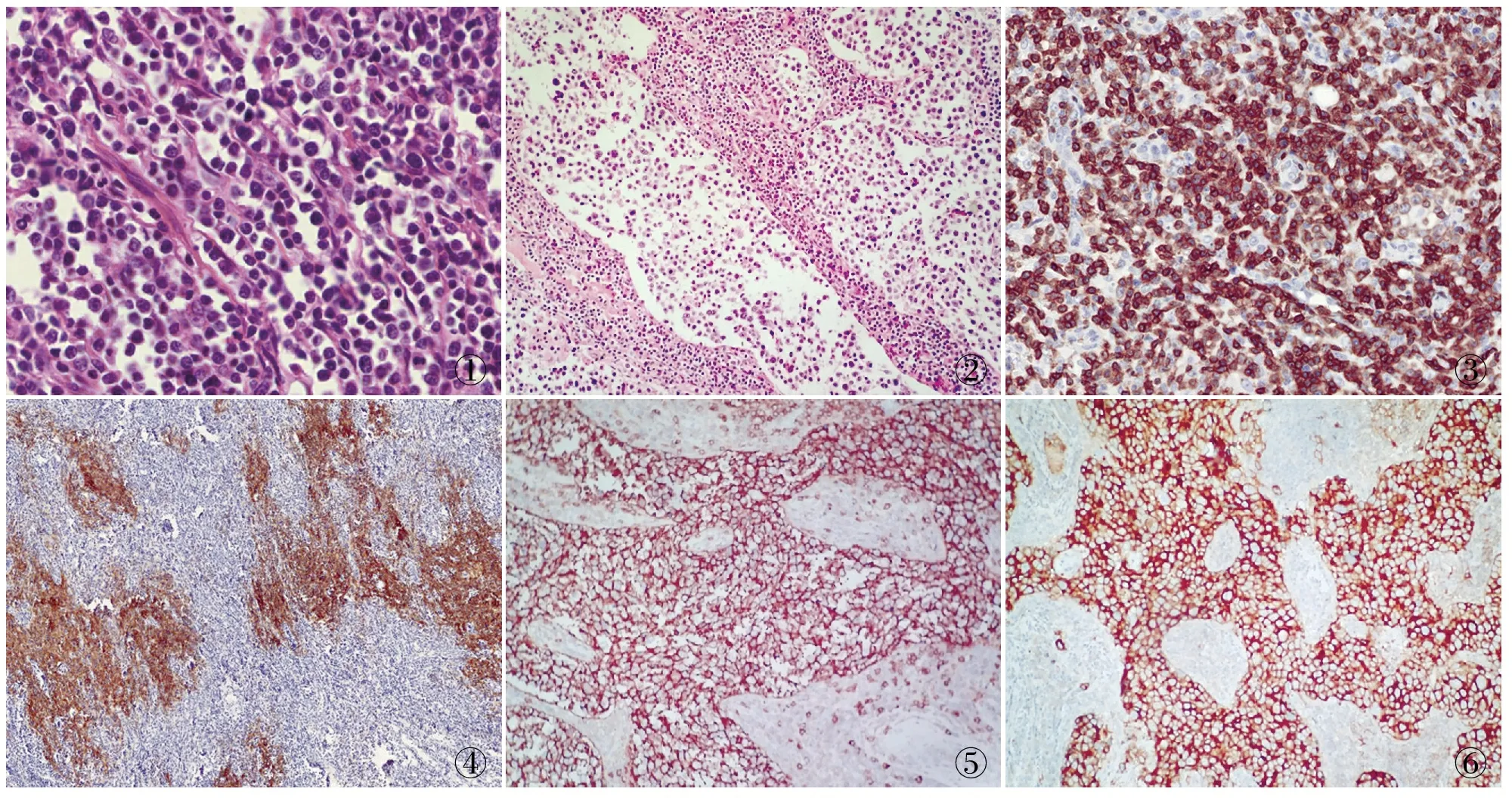
①②③④⑤⑥
图1 淋巴结内可见多形性的小到中等大小的淋巴细胞,胞质透明,并见血管增生 图2 扩张的淋巴窦呈裂隙状或不规则的分支吻合状;瘤细胞呈多边形或圆形,细胞质丰富,细胞核圆形、椭圆形或略呈多形性,有1~2个位置居中、显著的嗜酸性大核仁,并伴大量成熟浆细胞增生
图3 肿瘤细胞中CD3呈阳性,EliVision两步法 图4 增生的淋巴滤泡树突细胞中CD21呈阳性,EliVision两步法 图5 肿瘤细胞中CD30呈弥漫阳性,EliVision两步法 图6 肿瘤细胞中CD20呈弥漫阳性,EliVision两步法
2.7 随访 3例PPCL失访,其余7例随访时间5~18个月,平均15个月。3例PPCL经VAD方案治疗后1例达CR,2例达PR;最终3例PPCL死亡,确诊后存活时间4~13个月。另1例PPCL经沙利度胺联合VAD方案2个疗程治疗达CR,获得8个月CR后,复发,改用联合硼替佐米再次达CR,确诊后存活时间为18个月。CD30阳性的窦内弥漫大B细胞淋巴瘤伴反应性浆细胞增多症经R-CHOP方案化疗4个疗程后,各项实验室检查基本恢复正常,随访6个月,未复发;1例伴免疫性血小板减少性紫癜的血管免疫母细胞性T细胞淋巴瘤患者经CHOP方案1个疗程结束后,血小板恢复正常,6个疗程后完全缓解,随访5个月,未复发;另1例血管免疫母细胞性T细胞淋巴瘤经CHOP方案7个疗程后,缓解,随访7个月未复发。
3 讨论
PPCL是具有高度侵袭性的浆细胞疾病,以不良预后和对常规治疗的低反应率是其主要特征,骨髓检查可见浆细胞恶性增殖,在欧美淋巴瘤分型[3]和WHO(2008)造血与淋巴组织肿瘤分类[4]中,PCL属于浆细胞骨髓瘤(plasma cell myeloma, PCM)的临床变异型,被归入成熟B细胞肿瘤。本组7例PPCL起病急,以进行性贫血、血小板减少、发热、骨痛、肝脾及淋巴结肿大常见,中位年龄56岁,临床特点不同于PCM而类似于急性白细胞的表现,与PCM相比其发病中位年龄更为年轻,淋巴结、肝脾肿大和肾功能衰竭更为常见,而溶骨性病变及骨痛少于PCM[5]。有研究显示白细胞总数可以正常,但通常增高,有时可高达100×109/L,80%的PCL临床表现为贫血,50%的病例血小板减低[6-7],本组7例PPCL有6例白细胞增高及血小板减低,均有贫血;有7例β2MG增高及6例LDH增高,6例Bun和7例CREA增高,显示肾功能不全,与相关文献报道基本一致。文献报道PPCL免疫球蛋白分型IgG(33%)、IgA(20%)、IgD(3%)、IgE(1%),35%患者表现为重链缺失(仅分泌轻链),小于10%患者为无分泌型[8]。本组7例PPCL中6例IgG(IgGΚ 5例、IgGλ 1例),1例IgAΚ。
白血病性浆细胞形态学特点包括PCM形态学谱的大多数表现,但也有部分浆细胞体积较小,胞质相对较少,类似与淋巴样浆细胞。国外报道PPCL骨髓中浆细胞可高达80%以上[9],形态呈多样性,与骨髓瘤细胞形态相似,但分化程度低的原始、幼稚浆细胞更多见[10-11]。本实验7例PPCL及2例血管免疫母细胞性T细胞淋巴瘤和1例CD30阳性的窦内弥漫大B细胞淋巴瘤均无明确的PCM病史,起病时外周血浆细胞比例均>20%,且均伴形态学异常,临床特征类似于急性白血病表现。因此在形态学上拟诊为PPCL,但从10例患者的骨髓及外周血浆细胞比例分析,7例PPCL的原、幼浆细胞的比例明显高于3例反应性的浆细胞增多症的比例。
本组在结合骨髓及外周血细胞形态基础上进行血清免疫固定电泳及外周血流式细胞术检测,发现7例存在单克隆免疫球蛋白,其中6例IgG(IgGΚ 5例、IgGλ 1例),1例IgAΚ;其余3例虽然也存在免疫球蛋白IgG明显增高现象,但均属于多克隆增高;流式细胞术检测发现10例外周血浆细胞的免疫表型共同特点为均表达浆细胞标记CD38及CD138,有7例轻链呈单克隆限制性表达,6例为Kappa限制性表达,1例为Lamda限制性表达,与血清免疫固定电泳检测结果一致;其他3例轻链未见限制性表达,因此考虑为反应性浆细胞增多症。PPCL与多发性骨髓瘤有类似的免疫表型,但PPCL中CD20呈强表达的比例高于多发性骨髓瘤,而多发性骨髓瘤更多表达CD56、CD117和HLA-DR。CD20阳性常预示患者生存期较短,CD56阳性常伴较好的预后,而CD56阴性的多发性骨髓瘤通常有髓外累及[12];本组7例PPCL有2例CD20呈阳性;林阳等[13]报道CD28有助于PPCL与SPCL的鉴别,PPCL的表达频率明显低于SPCL。本组10例患者外周血浆细胞比例均超过20%,且形态上有一定异型性,由此可见单纯依据形态学标准诊断PPCL易误诊,免疫组化标记对细胞上固有标志(抗原、分子或受体)的识别,既客观,可重复性又好。因此,我们认为形态学结合临床病理特征及免疫表型,可提高诊断PPCL的准确度。
反应性浆细胞增多症是指一组由多种原因或原发疾病引起的以骨髓成熟浆细胞增多为临床的综合征,临床表现多与原发疾病有关,其浆细胞的本质是良性的,诊断标准是骨髓中浆细胞大于或等于3%[1];近年来恶性肿瘤疾病引起免疫球蛋白增多、消瘦、贫血伴反应性浆细胞增多在临床上已逐渐引起重视。本组3例淋巴瘤引起的浆细胞增多与PPCL有类似的临床特征,尤其是在骨髓及外周血细胞形态学上有相似特征,单从形态学上是难以区分。3例浆细胞增多在外周血中的浆细胞数量以CD30阳性的窦内弥漫大B细胞淋巴瘤占有核细胞的比例最高,且形态学上更为幼稚,血清免疫球蛋白的含量最高;单凭形态学极易误诊为PPCL,需结合免疫固定电泳及其免疫表型鉴别是恶性克隆性浆细胞还是反应性浆细胞;CD30阳性的窦内弥漫大B细胞淋巴瘤在WHO(2008)造血与淋巴组织肿瘤分类中定义为非特殊类型的弥漫大B细胞淋巴瘤形态变异类型。该大B细胞淋巴瘤十分罕见,国内仅见少数个案报道[14-15],其组织形态学特征、免疫表型及预后与本组报道一致;但CD30阳性的窦内弥漫大B细胞淋巴瘤伴外周血浆细胞明显增多的病例尚未见相关报道。
血管免疫母细胞性T细胞淋巴瘤是一种系统性淋巴组织增殖性疾病,以多发淋巴结及肝脾肿大、出现皮疹、贫血以及多克隆性高γ球蛋白血症为特点;其组织学改变为淋巴结结构破坏,浸润细胞含有多种类型,淋巴细胞、浆细胞、嗜酸粒细胞、组织细胞和免疫母细胞,特征为高内皮小静脉的分支状增生和滤泡树突细胞弥漫增生,滤泡和生发中心常消失[16]。血管免疫母细胞性T细胞淋巴瘤细胞为成熟的CD4阳性辅助性T细胞,强表达CXCL-13因子,该因子上调可刺激高内皮静脉捕获B细胞,动员B细胞进入淋巴结生发中心,促进B细胞扩增、成熟、分化为浆细胞[17],且浆细胞是肿瘤中的反应性成分,临床常常合并多克隆高丙种球蛋白血症[18]。张雯洁等[19]报道1例血管免疫母细胞性T细胞淋巴瘤伴反应性浆细胞增多症,其浆细胞在骨髓中仅占10.5%,且外周血未见浆细胞,本组2例血管免疫母细胞性T细胞淋巴瘤伴浆细胞增多症,其浆细胞比例在外周血均大于20%,且有1例伴原发病相关的免疫性血小板减少性紫癜,该现象较为少见;无论从临床症状还是形态学表现,本组2例血管免疫母细胞性T细胞淋巴瘤伴反应性浆细胞增多症与PPCL均十分相似。因此,在缺乏免疫组化检测或免疫电泳检测时易误诊为PPCL。
综上所述,PPCL与淋巴瘤伴浆细胞增多症患者有相同临床表现及相似的血细胞形态学特征。因此,单纯依据细胞形态学标准诊断PPCL易误诊,对PPCL可进行流式细胞免疫表型分析及免疫组化确诊,对淋巴瘤伴浆细胞增多症的患者必需结合淋巴结组织学检查确诊。
[1] 张之南,沈 悌. 血液病诊断及疗效标准[M]. 3版. 北京: 科学出版社, 2007:139-143.
[2] Garcia-Sanz R, Orfao A, Gonzalez M,etal. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics[J]. Blood, 1999,93(3):1032-1037.
[3] Harris N L, Jaffe E S, Stein H,etal. A revised European-American classification of lymphoid-neoplasms: a proposal from the International Lymphoma Study Group[J]. Blood, 1994,84(5):1361-1392.
[4] Mckenna R W, Kyle R A, Kuehl W M,etal. Plasma cell myeloma[M]//WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008:202-208.
[5] Tiedemann R E, Gonzalez-Paz N, Kyle R A,etal. Genetic aberrations and survival in plasma cell leukemia[J]. Leukemia, 2008,22(5):1044-1052.
[6] Kosmo M A, Gale R P. Plasma cell leukemia[J]. Semin Hematol, 1987,24:202-208.
[7] Noel P, Kyle R A. Plasma cell leukemia: an evaluation of response to therapy[J]. Am J Med, 1987,83(6):1062-1068.
[8] Albarracin F, Fonseca R. Plasma cell leukemia[J]. Blood Rev, 2011,25(3):107-111.
[9] Pagano L, Valentini C G, De Stefano V,etal. Primary plasma cell leukemia: a retros-pective multicenter study of 73 patients[J]. Ann Oncol, 2011,22(3):1-8.
[10] D′Arena G, Valentini C G, Pietrantuono G,etal. Frontline chemotherapy with tezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA multiple myeloma working party[J]. Ann Oncol, 2012,23(6):1499-1502.
[11] 范立权,熊树民. 浆细胞白血病实验室诊断与鉴别的分析[J]. 诊断学理论与实践, 2008,7(4):412-415.
[12] García-Sanz R, Orfão A, González M,etal. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics[J]. Blood, 1999,93(3):1032-1037.
[13] 林 阳,万岁桂. 流式细胞术免疫表型分析在多发性骨髓瘤及其他浆细胞疾病诊断中的应用[J]. 标记免疫分析与临床, 2013,20(1):60-62.
[14] 李小秋,陆洪芬,杨 践,等. CD30阳性的窦性大B细胞淋巴瘤临床病理学分析[J]. 中华病理学杂志, 2002,31(4):305-308.
[15] 张晓娟,石劲松,王林娜,等. CD30阳性的窦性大B细胞淋巴瘤1例[J]. 临床与实验病理学杂志, 2015,31(7):835-836.
[16] Dogan A, Gaulard P, Jaffe E S,etal. Angioimmunoblastic T-cell lymphoma[M]//Swerdlow S H, Campo E, Jaffe E S,etal. World Health Organization classificaition of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues[M]. Lyon: IARC Press, 2008:309-311.
[17] 杨 雷,董晓燕,刘春水,等. 高丙种球蛋白血症的血管免疫母细胞淋巴瘤1例报告[J]. 中国实验诊断学, 2009,15(3):541-542.
[18] 李 权,盐 城. 血管免疫母细胞性T细胞淋巴瘤临床病理观察[J]. 中外医疗, 2012,31(30):35-36.
[19] 张雯洁,孙延庆,赵英玲,等. 血管免疫母细胞性T细胞淋巴瘤伴浆细胞增1例[J]. 中国肿瘤临床, 2014,(14):946-941.
Diagnostic and differential diagnostic of primary plasma cellleukemia and lymphoma with increased plasma cell
ZHANG Hui-chao, HUANG Chen, WANG Peng-yu, LI Hong, CHEN Yan-ning, ZHANG Hong, DING Ya-wen, GAO She-jun
(DepartmentofHematologyLaboratory,theFourthHospitalofHebeiMedicalUniversity,Shijiazhuang050011,China)
Purpose To investigate the diagnosis, differential diagnosis and clinical manifestation of primary plasma cell leukemia (PPCL) and lymphoma with increased plasma cell. Methods Through clinical data and cell morphology, flow cytometry (FCM), immunofixation electrophoresis and immunohistochemistry of EliVision two-step examination were used to analyze 7 cases of PPCL and 3 cases of lymphoma with increased plasma cell. Results All patients with PPCL and lymphoma with increased plasma cell presented with anemia, thrombocytopenia, fever, liver and spleen and lymph node swelling. The proportion of plasma cells in peripheral blood morphology were larger than 20%, accompanied by morphological abnormality. FCM of peripheral blood showed all 7 cases of PPCL expressed CD38 and CD138, CD56 expression in the 2 cases and CD20 in the 2 cases. The light chain (Lamda, Kappa) showed a monoclonal restricted expression, which was consistent with the diagnosis of PPCL. CD19 and CD45 were weakly positive in 3 cases of lymphoma with increased plasma cell, CD38 and CD138 were positive, and no restricted expression was found in light chain IgL, wich belonging to the immunophenotypes of normal plasma cells. Of 3 cases of light chain (Ig) without restrictive expression, 2 of them were angioimmunoblastic T-cell lymphoma (ATCL) and 1 case was CD30-positive sinusoidal large B-cell lymphoma (CD30+SLBCL) that confirmed by lymph node biopsy and pathological examination. Conclusion The PPCL and lymphoma with increased plasma cell have the same clinical manifestations and similar morphological characteristics of blood cells. The diagnosis of PPCL should be combined with immunoelectrophoresis and FCM, and the diagnosis of lymphoma with increased plasma cell needs to be confirmed by histological examination of lymph nodes.
primary plasma cell leukemia; lymphoma; reactive plasmacytosis; immunohistochemistry
河北省科技厅大健康服务和生物医药专项课题(162777243)
河北医科大学第四医院血液病实验室,石家庄 050011
张会超,男,硕士,主管检验师。E-mail:zhanghuichao050108@163.com 高社军,男,主任技师,通讯作者。Tel:(0311)66696373,E-mail:gaoshe@sina.com.cn
时间:2017-5-17 23:53 网络出版地址:http://kns.cnki.net/kcms/detail/34.1073.R.20170517.2352.008.html
R 446;R 392.31
A
1001-7399(2017)05-0505-06
10.13315/j.cnki.cjcep.2017.05.008
接受日期:2017-03-17
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:22
中国临床医学影像杂志(2021年5期)2021-08-13 09:01:38
天津医科大学学报(2021年3期)2021-07-21 09:04:00
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
家庭医学(下半月)(2019年11期)2020-01-16 08:39:06
装备制造技术(2019年12期)2019-12-25 03:07:10
中国生殖健康(2019年9期)2019-01-07 01:19:00
上海建材(2017年4期)2017-04-06 07:32:03
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
