
猪细环病毒数字PCR定量检测方法的建立
2017-06-01 10:40杨春华罗秋红祝建新孙思扬
中国动物检疫 2017年4期
杨春华,孙 洁,罗秋红,祝建新,孙思扬,周 延,钟 毅,夏 彪
(1. 江西出入境检验检疫局,江西南昌 330002;2. 深圳出入境检验检疫局,广东深圳 518045;3. 南昌大学,江西南昌 330002)
猪细环病毒数字PCR定量检测方法的建立
杨春华1,孙 洁2,罗秋红1,祝建新1,孙思扬1,周 延3,钟 毅1,夏 彪1
(1. 江西出入境检验检疫局,江西南昌 330002;2. 深圳出入境检验检疫局,广东深圳 518045;3. 南昌大学,江西南昌 330002)
[目的]实现猪细环病毒(TTSuV)的准确定量检测。[方法]根据TTSuV的序列特点,设计特异性引物、探针,建立数字PCR检测技术。对数字PCR反应体系中的引物和探针浓度进行优化,分析方法的灵敏度、特异性,并初步应用于进行临床检测。[结果]最终确定TTSuV1a和TTSuV1b数字PCR反应体系中最佳引物浓度均为250 nmol/L,最佳探针浓度均为 300 nmol/L,TTSuV1a型和TTSuV1b型灵敏度均可达到单个拷贝数;以猪圆环病毒Ⅱ型、猪细小病毒和猪伪狂犬病毒进行特异性试验,结果无交叉反应;批内和批间试验表明,该方法的重复性良好;本实验室留存的92份血清样本的检测结果与其背景信息一致。[结论]本研究建立的TTSuV数字PCR法具有特异性强、灵敏度高、检测限低等优点,可用于TTSuV的定量检测。
猪细环病毒;实时荧光PCR;数字PCR
细环病毒(Torque Torque Virus,TTV)是1997 年首次发现的,可感染人[1-2]以及猪、牛、羊、猫、狗等多种动物[3-4]的一种病毒。1999年Leary首次报道了猪细环病毒(Torque Torque Sus Virus,TTSuV)的存在[3]。该病毒是单股、负链、环状DNA 病毒[3,5],无囊膜,在氯化铯(CsCl)溶液内的浮密度为1.31~1.35 g/cm3,在粪便、血液、胆汁中的浮密度也大致与其相同。TTSuV的基因组长度约为3.9 kb,具有ORF1 和ORF2两个开放阅读框,其中ORF1编码病毒的衣壳蛋白,ORF2编码病毒的非结构蛋白[6]。2011年国际病毒分类委员会(ICTV)将TTSuV重新进行了分类,分为Torque Torque Sus Virus 1和Torque Torque Sus Virus K2,其中常引起猪只感染的Torque Torque Sus Virus 1又分 为Torque Torque Sus Virus 1a和Torque Torque Sus Virus 1b。
目前,关于TTSuV的检测方法主要有酶联免疫吸附试验法(ELISA)、免疫印迹法、PCR凝胶电泳法和实时荧光PCR方法等[7-9]。ELISA法和免疫印迹法操作复杂、费时、特异性不强。实时荧光PCR是目前进行病毒核酸检测的主流技术手段。随着科技的发展,在实时荧光PCR的基础上,发展出了一种核酸检测绝对定量的新技术,即数字PCR技术。同传统PCR以及qPCR相比,数字PCR增加了一步分液的操作,将几十微升的反应体系分成上百万个微小的、独立的反应体系。核酸模板在这种分液的过程中被充分稀释,理想状态下每个小反应体系中至多含有1个分子的核酸模板。分液完成后,收集所有液滴进行常规PCR扩增,扩增完成后识别荧光信号并计数,从而得到绝对定量结果。与qPCR不同,数字PCR技术无需设置外部核酸标准,可实现核酸模板的绝对定量分析,在病原体基因检测方面具有广阔的应用前景。本研究旨在根据TTSuV的核酸序列,建立特异性强、敏感性高的TTSuV数字PCR检测技术。
1 材料与方法
1.1 样品采集
从江西省的4个规模猪场,采取猪血清样本92份。
1.2 仪器和试剂
1.2.1 主要仪器。核酸抽提纯化系统(MagNA Pure LC2.0,瑞士Roche公司);Nanodrop 3300超微量紫外分光光度计(美国赛默飞世尔公司);实时荧光PCR扩增仪(Biorad CFX96,美国Biorad公司);基因扩增仪(ABI9700,美国ABI公司);Raindrop数字PCR仪(美国RainDance Technologies公司)。
1.2.2 主要试剂。病毒DNA提取试剂盒(MagNA Pure LC总核酸分离试剂盒);实时荧光PCR试剂盒(美国Life Technology公司);数字PCR试剂盒(美国RainDance Technologies公司); PicoGreen双链DNA检测试剂盒(美国RainDance Technologies公司)。所有引物、探针由大连宝生物公司合成。
1.2.3 引物设计。从GenBank下载TTSuV所有靶基因的序列,用Lasergene进行核酸序列比对,并截取其中最一致的序列,设计实时荧光PCR引物、探针,分别是针对TTSuV1a的Q-F1和Q-R1引物对及探针Q-P1,针对TTSuV1b的Q-F2和Q-R2引物对及探针Q-P2,并验证其保守性;根据实时荧光PCR引物所包含的核酸片段,选择克隆用引物,分别为针对TTSuV1a的引物S1、S2,针对TTSuV1b的引物T1、T2[10](表1)。

表1 引物序列
1.3 试验方法
1.3.1 病毒DNA的提取及标准质粒模板的制备
1.3.1.1 病毒DNA的提取。用核酸抽提纯化系统MagNA Pure LC2.0,并使用该设备配套的核酸提取试剂盒,按照试剂盒说明书提取样品总核酸,用60 µL TE缓冲液将其充分溶解,- 20 ℃保存备用。
1.3.1.2 目的基因的克隆、测序。以提取的病毒DNA为模板,分别用引物S1和S2、T1和T2进行TTSuV1a、TTSuV1b的全序列扩增,将PCR扩增产物经电泳及鉴定后,获取重组质粒,并进行PCR、酶切及测序验证,验证后的重组质粒分别命名为T-TTSuV1a和T-TTSuV1b。
1.3.1.3 质粒T-TTSuV1a和T-TTSuV1b的定量。用PicoGreen试剂盒中的DNA干粉标准品,按照说明书的要求配制成1 mg/mL的标准品工作液并进行倍比稀释,用ND3300紫外分光光度计检测荧光值,作直线回归,制备标准曲线。同时以1×TE缓冲液作为空白对照,测定质粒T-TTSuV1a和T-TTSuV1b及空白对照的荧光值,并根据标准曲线进行质粒浓度的计算。
1.3.2 数字PCR反应体系及扩增条件的优化。基于先进行PCR反应,再进行荧光检测的数字PCR的原理,在反应体系和反应条件的摸索过程中,先用qPCR验证引物、探针的可靠性,同时调整Taq酶、引物探针用量、循环参数和循环条件,使之达到最佳反应条件,再进行数字PCR反应体系和条件的微调。
1.3.3 数字PCR反应的灵敏度。将重组质粒T-TTSuV1a和T-TTSuV1b进行梯度稀释,分别标记为S0~S8,根据紫外分光光度计测定结果计算质粒浓度在总体积为20 µL的数字PCR反应体系中加入稀释好的S0~S8的DNA模板2 µL,使拷贝数分别为107、106、105、104、103、102、101、100数量级,进行灵敏度研究。数字PCR扩增的循环条件为:95 ℃预变性90 s;95 ℃变性10 s,55 ℃退火10 s,72 ℃延伸30 s,40个循环;72 ℃延伸7 min,4 ℃保存反应产物。
1.3.4 数字PCR反应的特异性试验。按照常规方法,利用Roche MagNA Pure LC 2.0,对猪伪狂犬病毒(Pseudorabies virus,PRV)、猪细小病毒(Porcine Parvovlirus,PPV)、猪圆环病毒Ⅱ型(Porcinecircovirus type2,PCV2)等病毒提取核酸,分别使用1.3.2的方法,对上述病毒的核酸进行检测,以确定该方法的特异性(分别以T-TTSuV1a和T-TTSuV1b为阳性对照,DEPC水为阴性对照)。
1.3.5 数字PCR反应的重复性试验。分别设置3个不同浓度的重组质粒T-TTSuV1a和T-TTSuV1b,应用1.3.2的方法,在同一反应条件下进行5次独立试验,以确定此方法的批内重复性;连续5天对相同的样品进行检测,以确定此方法的批间重复性。
1.3.6 临床样本检测的初步应用。将本研究建立的数字PCR检测方法应用于实验室检测,从江西省4个规模猪场采集血清样本92份,提取DNA,进行数字PCR检测。
2 结果
2.1 优化后的探针浓度及反应条件
采用qPCR对探针的浓度进行优化和确定,使TTSuV1a和TTSuV1b反应体系内的探针浓度分别 为50、100、150、200、250、300、350和400 nmol/L。当TTSuV1a探针浓度为250 nmol/L时,荧光信号强度较高,且不再随着浓度的增加而增加;TTSuV1b的最佳探针浓度为300 nmol/L。后续试验中,将TTSuV1a和TTSuV1b PCR反应的探针浓度分别设置为250 nmol/L和300 nmol/L。
采用qPCR对反应条件进行优化和确定。对T-TTSuV1a标准品10-1的稀释质粒,在53~65 ℃范围内的8个梯度退火温度,进行荧光定量PCR反应。结果显示:温度越高,扩增效率越低(图1);退火温度为55 ℃时扩增效率最高,E=101.9%。对TTSuV1b标准品10-1的稀释质粒,在53~65℃范围内的8个不同退火温度,进行荧光定量PCR反应。结果显示:温度越高,扩增效率越低(图2);退火温度为55 ℃时,扩增效率最高,E=97.5%。
T-TTSuV1a和TTSuV1b优化后的反应条件均为:95 ℃预变性90 s;95 ℃变性10 s,55 ℃退火10 s,72 ℃延伸30 s,40个循环;72 ℃延伸7 min,4 ℃保存反应产物。

图1 TTSuV1a标准品(10-1)在53~60 ℃范围内的实时荧光PCR反应结果

图2 TTSuV1b标准品(10-1)在53~60 ℃范围内的实时荧光PCR反应结果
应用优化后的反应体系和条件进行数字PCR试验,TTSuV1a和TTSuV1b扩增的散点图分别见图3、图4。经过优化的PCR体系对TTSuV的扩增效果良好,其中阳性微滴和阴性微滴明显分成两簇,而且中间弥散的微滴数目很少,表明经过优化确定的数字PCR探针浓度和扩增体系适合对TTSuV进行定量分析。
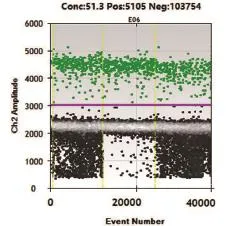
图3 微滴数字PCR方法扩增TTSuV1a的散点图
2.2 数字PCR反应的灵敏度
将TTSuV1a和TTSuV1b的DNA模板稀释后,进行数字PCR反应,反应体系S1~S8生成的可接受总微滴数和阳性微滴数分布见图5(TTSuV1a)和图6(TTSuV1b)。从图中可发现,DNA样品经稀释后再扩增,其阳性微滴数目随着浓度的降低而逐渐减少。在数字PCR反应中,当20 µL反应液中的拷贝数为100数量级时,TTSuV1a检测到5个扩增,TTSuV1b检测到4个扩增,且对照组均无扩增,说明数字PCR对TTSuV1a和TTSuV1b的检测灵敏度均可达到单个拷贝。

图4 微滴数字PCR方法扩增TTSuV1b的散点图

图5 数字PCR方法扩增TTSuV1a的灵敏度

图6 数字PCR方法扩增TTSuV1b的灵敏度
2.3 特异性试验
采用本实验建立的TTSuV1a和TTSuV1b 实时荧光PCR方法,对TTSuV1a和TTSuV1b及PPV、PCV-2和PRV 3种病毒培养液提取的DNA进行数字PCR方法特异性验证。结果表明,TTSuV1a和TTSuV1b均得到了特异性扩增,其他病毒的检测均为阴性,且TTSuV1a和TTSuV1b两者之间无交叉反应,表明建立的数字PCR方法特异性良好(图7、图8)。
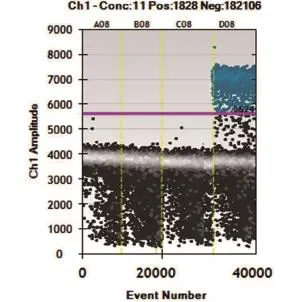
图7 数字PCR方法扩增TTSuV1a的特异性

图8 数字PCR方法扩增TTSuV1b的特异性
2.4 重复性试验
对TTSuV1a和TTSuV1进行数字PCR检测的批内和批间重复试验。分别取模板浓度为103、102、101的TTSuV1a和TTSuV1b质粒标准品,在同一反应条件下,分别做5个批内重复和批间重复。结果显示,TTSuV1a和TTSuV1b数字PCR方法批内重复变异系数均小于1%,批间重复变异系数均小于3%(表2、表3),表明方法重复性好。
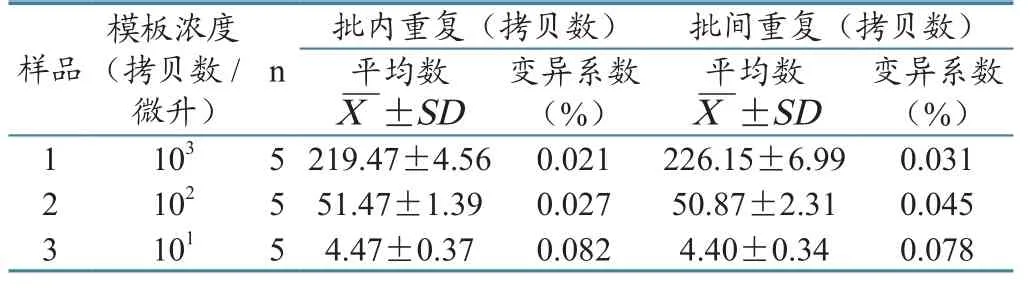
表2 数字PCR检测TTSuV1a批内及批间重复试验结果

表3 数字PCR检测TTSuV1b批内及批间重复试验结果
2.5 临床样品检测的初步应用
对92份临床血液样本提取核酸,采用本研究建立的数字PCR方法检测。结果表明,92份样品中,阳性样品份数为17,每个样品的检测结果均与其背景信息相吻合。
3 讨论
当前,动物疫病的主要检测手段以qPCR为主。qPCR检测是通过标准曲线来计算样品中含有的目标基因拷贝数的,依赖于Ct值的重现性、Ct值与起始模板的线性关系,其最终计算出的目标基因拷贝数是相对定量的。在日常动物疫病检测中,经常会出现当靶标序列含量很低时,qPCR的Ct值处于临界值附近,这时应用qPCR无法得到准确的结果,需要一种更加灵敏的方法来检测低含量的核酸,此时数字PCR技术应运而生。这是一种新兴的定量技术,其操作步骤是先进行分液,即将一定体积的PCR反应体系快速、均一地制备成数百万个皮升大小的油包水液滴,确保真正的单分子扩增。分液完成后,收集所有液滴进行常规的PCR扩增。与qPCR不同,数字PCR不对扩增过程进行实时监测,而是检测反应终点的荧光信号。对结果的判读是基于反应终点阅读到的荧光信号强度,结合无模板对照和阴阳性对照划出相应的阈值。将信号强度位于阈值之上的微滴判定为阳性微滴,位于阈值之下的判定为阴性微滴,并利用泊松分布统计学公式计算出目的DNA分子浓度。该技术在检测时不需构建标准曲线,即可直接得到绝对定量。当前,数字PCR技术被大力推广用于特定基因及核酸的绝对定量检测中,尤其是对微量样品的绝对定量。数字PCR以qPCR为基础,是对单个微滴中的目标基因进行检测,可直接计算出目标基因DNA分子的个数,是对起始样品的绝对定量,适用于需要绝对定量及qPCR中依赖Ct值不能很好分辨检测结果的情况。
数字PCR是一种绝对定量方法。数字PCR体系中的DNA浓度若过高,会导致微滴不满足泊松分布原理,使定量结果显著偏离真实值,无法实现数字PCR的准确定量。因此,DNA浓度的准确定量对于方法的建立十分关键。本文对质粒模板的浓度测定,未采用传统的OD260/280测量法,而是采用ND3300紫外分光光度计,结合PicoGreen双链DNA检测试剂盒进行。这是因为应用OD260/280测量核酸浓度时,单链核苷酸、单核苷酸和样本中的杂质都会对结果产生干扰,同时光吸收不能区分DNA和RNA,但PicoGreen染料法能够排除上述干扰,使得测量的DNA浓度更加准确。本文选取了猪细环病毒DNA序列的保守区设计引物探针,并通过qPCR反应测试其特异性,在后续的数字PCR试验中,NTC均没有检测到阳性微滴,可见该体系中无污染,无非特异性扩增,方法的特异性良好,经反复调整反应体系确定其最佳浓度,以此来摸索数字PCR的最佳反应条件和反应体系。
过去,对猪细环病毒的研究主要集中在检测方法及感染率的调查等方面[10-11]。近年来,随着对其研究的深入,对检测方法的要求也越来越高,逐渐出现了实时荧光PCR法、PCR-DHPLC等[12-13]。本研究在实时荧光PCR方法的基础上,将数字PCR方法引入到该病毒的检测中,建立了数字PCR检测方法,能够实现对猪细环病毒的准确定量检测,且该方法灵敏度高、特异性强、使用方便,可在短时间内出具定量结果,实现了痕量病原检测,适用于对猪细环病毒早期感染的鉴定,对猪细环病毒的致病性及机理的进一步研究具有重要意义。
[1]NISHIZAWA T,OKAMOTO H,KONISHI K,et al. A novel DNA virus(TTV)associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology [J]. Biochem Biophys Res Commun,1997,241:92-97.
[2]庄辉.TT病毒的研究现状及展望[J]. 中华内科杂志,1999,38(11):727-728.
[3]OKAMOTO H,TAKAHASHI M,NISHIZAWA T,et al. Genomic characterization of TT viruses(TTVs)in pigs,cats and dogs and their relatedness with species-specific TTVs in primates and tupaias [J]. J Gen Virol,2002,83:1291-1297.
[4]ZHENG H,YE L,FANG X,et al. Torque teno virus(SANBAN isolate)ORF2 protein suppresses NF-kappa B pathways via interaction with IkappaB kinases [J]. J Virol,2007,81:11917-11924.
[5]VILLIERS E D,HAUSEN H Z. Classification of TTV and related viruses(Anelloviruses)[J]. Current Topics in Microbiology and Immunology,2009,331:21-33.
[6]OKAMOTO H,NISHIZAWA T,KATO N,et al. Molecular cloning and characterization of a noval DNA virus(TTV)associated with posttransfusion hepatitis of unknown etiology[J]. Hepatol Res,1998,10:1-16.
[7]李阳,肖洁. TTV 检测方法研究进展[J]. 重庆医学,2006,35(18):1718-1720.
[8]赵焕英,包金风. 实时荧光定量PCR技术的原理及其应用研究进展[J]. 中国组织化学与细胞化学杂志,2007,16(4):492-497.
[9]KEKARAINEN T,LOPEZSORIA S,SEGALE J. Detection of swine Torque teno virus genogroups 1 and 2 in boar sera and semen [J]. Theriogenology,2007,68:966-971.
[10]田光玲,何后军,张黎,等. 江西省猪细环病毒1型和2型流行情况调查及全基因组克隆与序列分析[J]. 中国兽医学报,2016,36(7):1098-1102.
[11]SEGALE J,MARTINEZ G L,CORTEY M,et al. Retrospective study on swine Torque teno virus genogroups 1 and 2 infection from 1985 to 2005 in Spain [J]. Veterinary Microbiology,2009,134:199-207.
[12]杨春华,周延,孙思扬,等. 猪细环病毒 PCR-DHPLC检测技术的建立及应用[J]. 畜牧兽医学报,2012,43(9):1422-1428.
[13]BRASSARD J,GAGEN M J,HOUDE A,et al. Development of a real-time TaqMan PCR assay for the detection of porcine and bovine Torque teno virus [J]. J Applied Microbiology,2010,108:2191-2198.
(责任编辑:朱迪国)
Development of a Digital PCR Assay for Quantitative Detection of Torque Teno Sus Virus
Yang Chunhua1,Sun Jie2,Luo Qiuhong1,Zhu Jianxin1,Sun Siyang1,Zhou Yan3,Zhong Yi,Xia Biao1
(1. Jiangxi Entry-exit Inspection & Quarantine Bureau,Nanchang,Jiangxi 330002;2. Shenzhen Entry-exit Inspection & Quarantine Burea,Shenzhen,Guangdong 518045;3. Nanchang University,Nanchang,Jiangxi 330002)
[Objective] In order to accurately and quantitatively detect the Torque Teno Sus virus(TTSuV). [Methods] Specific primers and probes were designed and synthesized based on the sequence of TTSuV,then a digital PCR detection assay(dPCR)was established. After optimizing the working concentration of primers and probes,the sensitivity and specificity of the established reaction system were tested,and preliminary application was carried out. [Results] The optimal concentration of primers for both TTSuV1a and TTSuV1b genes had the same value of 250 nmol/L,and the optimal concentration of probes for the two genes was 300 nmol/L. As to the sensitivity of dPCR method,results showed single copy of TTSuV1a and TTSuV1b could be detected. Then its specificitywas evaluated by detecting TTSuV,Parvovirus(PPV),Porcine circovirus type 2(PCV-2)and Pseudorabies virus(PRV)simultaneously,and no cross reaction was observed. Furthermore,good repeatability of the method was confirmed by intra and inter assay. At last,92 clinical serum samples derived from our laboratory were conducted detection,results of which showed good consistence with their background information. [Conclusion]The established dPCR method was specific,sensitive and repeatable.,and it could be used in quantitative detection of TTSuV.
TTSuV;real-time PCR;digital PCR
S851.34
A
1005-944X(2017)04-0080-07
10.3969/j.issn.1005-944X.2017.04.024
江西省科技计划项目(20151BBF60048)
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
计测技术(2022年1期)2022-04-18
快乐语文(2021年34期)2022-01-18
食品安全导刊(2021年21期)2021-08-30
中国(俄文)(2020年8期)2020-11-23
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国外汇(2019年22期)2019-05-21
北京航空航天大学学报(2017年2期)2017-11-24
兵工学报(2012年8期)2012-02-23
