
Role of Cholesterol Crystals During Acute Myocardial Infarction and Cerebrovascular Accident
2017-05-23 08:44:02JagadeeshKalavakuntaMDMayankMittalMDAbedJanoudiPhDOliverAbelaMDFadiAlreefiMDandGeorgeAbelaMScMD
Jagadeesh K. Kalavakunta, MD, Mayank K. Mittal, MD, Abed Janoudi, PhD,Oliver G. Abela, MD, Fadi Alreefi, MD and George S. Abela, MSc, MD
1Department of Cardiology, Michigan State University/Borgess Medical Center, Kalamazoo, MI, USA
2Department of Medicine/Cardiology, Michigan State University, East Lansing, MI, USA
3Department of Medicine/Cardiology, University of Nevada, Las Vegas, USA
Introduction
Ischemic heart disease and stroke are the leading global causes of morbidity and death [1, 2]. Although the pathophysiology of these conditions has been investigated extensively, much has yet to be discovered. In this review we will explore the role of cholesterol crystals (CCs) in acute myocardial infarction (AMI) and cerebrovascular accident (CVA).Arterial occlusion, either partial or complete, can cause acute coronary syndrome (ACS) or stroke. The pioneering efforts of Constatinides [3] and Davies[4] have led to the understanding that plaque rupture and erosion are key causes of arterial obstruction that causes AMI or CVA. Many hypotheses have been proposed for the mechanism of plaque rupture leading to arterial thrombosis. These include hemorrhage into the plaque from the vasa vasorum, arterial vasospasm, and inflammation by macrophages releasing metalloproteinases that can digest the fibrous cap[5–7]. All these mechanisms seem to contribute to plaque rupture at various stages of development of the vulnerable plaque but no evidence has been provided of a direct causal relationship between these concepts and plaque rupture. However, recent work has provided evidence that cholesterol expands to occupy greater volume when it crystalizes from a liquid to a solid state [8, 9]. The presence of extensive CCs in many atherosclerotic plaques raises the question as to their potential role in plaque rupture and erosion.
Vulnerable Plaque to Eventual Rupture
Atherosclerotic plaque formation begins with the entry of low-density lipoprotein (LDL) particles into the subintimal space, initiating a cascade of inflammatory changes through activated macrophages,mast cells, and T lymphocytes [10]. These reactions ultimately lead to positive remodeling of the arterial wall and the development of vulnerable plaque,which is physically unstable [11]. The histologic characteristics of the vulnerable plaque have been defined by pathology as a plaque with a large lipidrich necrotic core (LRNC) covered by a thin fibrous cap, a reduced number of structural cells, and in filtration with inflammatory cells [12, 13]. This is typically the lesion that is found in association with rupture and arterial thrombosis that ultimately leads to clinical events, such as ACS [14] and CVA.However, there remains a paucity of data regarding the sentinel event known as plaque rupture. Through a series of in vitro and autopsy studies, our group has added to the growing body of evidence elucidating the role of subintimal CCs. This includes the role CCs in vascular inflammation and mechanical injury to the fibrous cap leading to rupture by crystallization and volume expansion [8, 9, 15].
Two stages are described as the steps leading from inflammation to mechanical plaque rupture [15].
Stage 1 is characterized by CC-induced foam cell apoptosis [16] and cellular injury that also recruits macrophages. These are believed to propagate cytokine- and chemokine-induced local inflammation, ultimately forming the LRNC that is a critical element of the vulnerable plaque.
Stage 2 is characterized by CC-induced rupture of plaque and arterial wall injury that leads to atherosclerosis-mediated clinical syndromes and systemic inflammation. The CCs can cause arterial wall damage through a combination of physical and chemical changes occurring in the vessel wall. Scanning electron microscopy (SEM) of culprit human coronary arteries involved in AMI demonstrated CCs piercing the intimal surface and fibrous cap. The CCs have sharp edges that can cause direct injury to the arterial wall, as was demonstrated in benchtop experiments and con firmed in coronary arteries from patients who died of AMI [8, 15, 17–19] (Figures 1 and 2). However, patients who died of noncardiogenic causes but also had significant atherosclerosis in their coronary arteries did not have evidence of CCs perforating through the intimal surface. This lends support to the concept that crystallization leading to rupture is a major underlying mechanism for ACS. Also, other investigators using a different technique with micro optical CT have examined atherosclerotic plaques and demonstrated the presence of CCs perforating the intimal fibrous cap [20]. More recent work with optical CT during coronary interventions in humans has demonstrated presence of CCs in the arterial wall plaque at sites of disrupted plaques [21, 22]. CC expansion leading to rupture is enhanced by the local physicochemical changes of the milieu surrounding the plaque and arterial wall [23]. More importantly,the release of large quantities of CCs into the arterial lumen has the potential of occluding the vessel independently of thrombosis or vasospasm [8, 9].
Cholesterol Crystals Triggers Inflammation in Atherosclerotic Plaques
At Michigan State University, investigators originally proposed that CCs can trigger inflammation in atherosclerotic plaques similar to the inflammatory effects of monosodium urate crystals in gout [15, 24]. This concept was then con firmed in subsequent studies by Düwell et al. [25] and others [26] demonstrating that CCs can trigger an inflammatory response in an atherosclerotic mouse model via the nucleotide-binding domain, leucine-rich repeat, and pyrin domain–containing 3 (NLRP3) inflammasome similarly to urate crystals [27]. The NLRP3 inflammasome activates IL-1β, which cascades to activate IL-6 and causes the liver to produce C-reactive protein, which has been proposed by Ridker [28] as a biomarker associated with increased risk of cardiovascular events.

Figure 1 Demonstration of Volume Expansion of Cholesterol During Crystallization.
Cholesterol Crystals Trigger Mechanical Injury of Atherosclerotic Plaque
In a series of in vitro experiments, Abela and Aziz [8,9] studied the crystallization of cholesterol powder in graduated cylinders and measured the change in volume when cholesterol crystallizes. Cholesterol crystallization was associated with an up to 45% increase in peak volume in 3 min, and the tips of these CCs were sharp enough cut and tear 10–40-µm-thick fibrous membranes. They also showed a statistically significant relationship between the amount of cholesterol and the peak level (r=0.98,P<0.01) and the rate of CC growth (r=0.99,P<0.01) (Figure 1).This has implications regarding the large size of the LRNC, and an increased risk of rupture was observed by Loree et al. [29]. These results were reproduced in a separate in vitro model that studied the impact of local physical factors on cholesterol crystallization on volume expansion [23]. In those studies, the researchers added various amounts of cholesterol powder to graduated cylinders and dissolved them in corn oil and demonstrated a similar phenomenon of crystallization at body temperature (37 °C). Increased cholesterol crystallization and volume expansion,thought to also occur in vulnerable plaque, was associated with increased saturation of cholesterol. The increase in volume of cholesterol during crystallization was 0.05 mL at 44 °C compared with 0.5 mL at 22 °C. Lowering the temperature not only increased volume expansion but also initiated crystallization earlier. Also, rising pH and hydration of the cholesterol molecule to the monohydrate form were factors that increased volume expansion. Cholesterol monohydrate had been previously reported as the predominant species in human plaques [30]. Moreover,changes in temperature and saturation had a synergistic effect on volume expansion. The influence of temperature could help explain the increase in early morning cardiovascular events since core temperature is lower at that time compared with later times of the day [31]. Increased cholesterol crystallization at colder temperature may at least in part also explain the increase in the incidence of cardiovascular events during the winter season. This increase in the incidence of ACS during colder weather has been observed worldwide and noted especially during snow shoveling activity [32, 33].
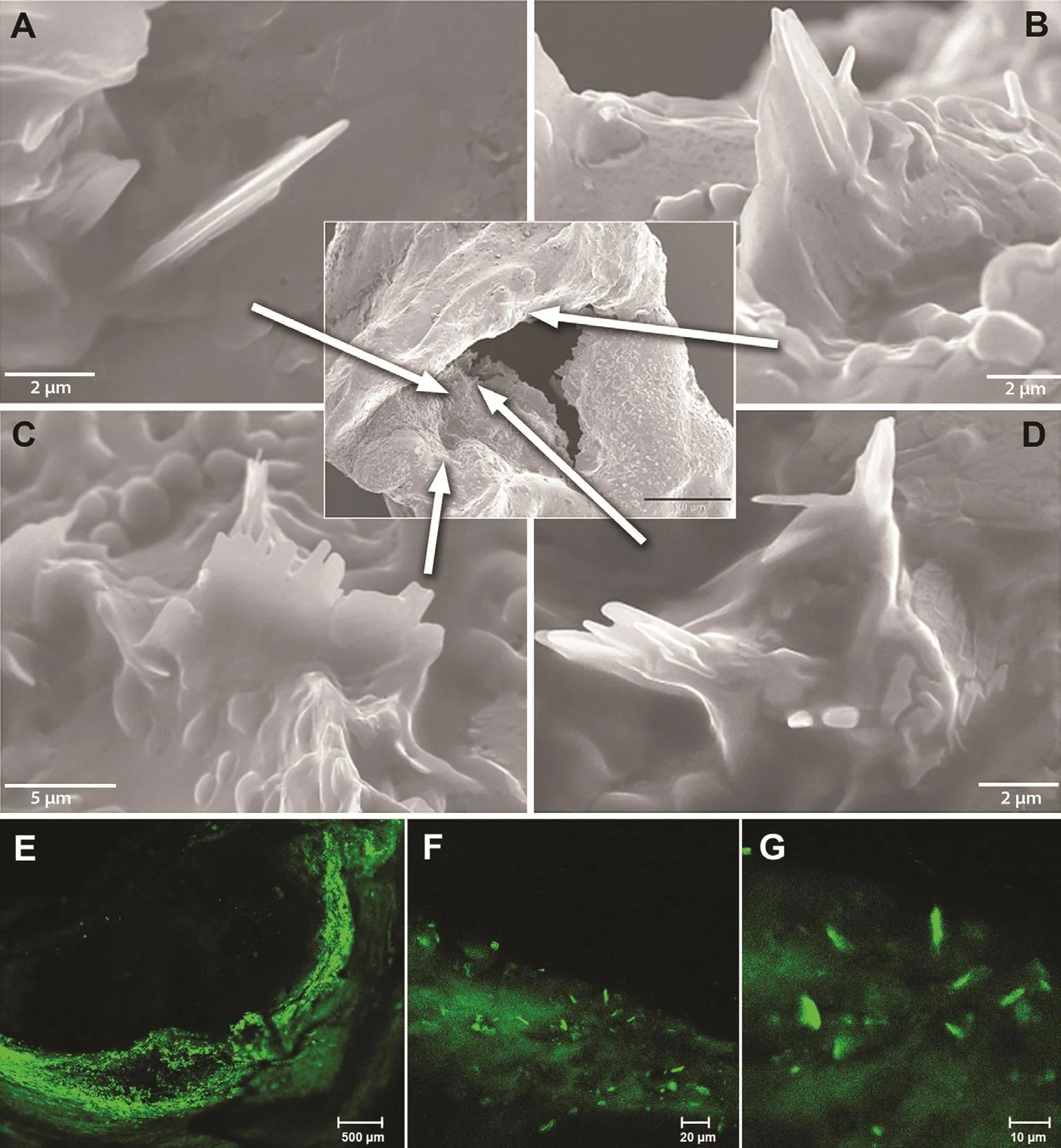
Figure 2 Scanning Electron Micrographs of Human Coronary Artery and Fluorescence Microscopy of Carotid Plaque.
In the past the amount and role of CCs have been greatly under appreciated and unrecognized because of the tissue preparation techniques, which use high concentrations of ethanol to dehydrate the tissue.This process readily dissolves the CCs, which then are noted as “clefts” by light microscopy [34].However, by avoidance of the use of ethanol and with the use of either air or a vacuum to dehydrate tissues, it is possible to detect and quantify the extent of CCs [19] (Figure 3).
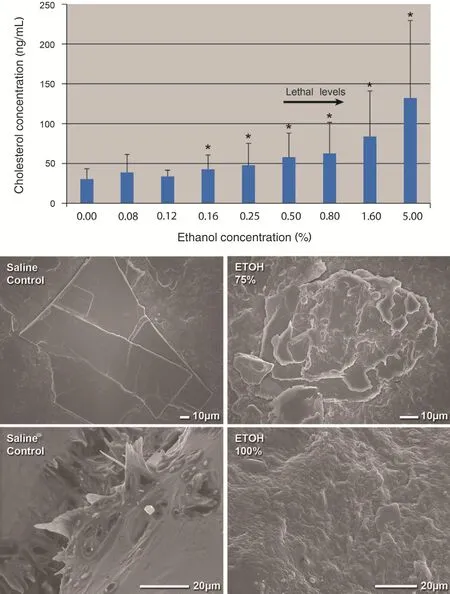
Figure 3 Solubility of Cholesterol Crystals and Scanning Electron Micrographs of Carotid Plaques at Various Ethanol Concentrations.
In a recent study, our group demonstrated that ethanol dissolves CCs even at concentrations that are within physiologically tolerated serum levels [19].However, the high concentrations of ethanol used to dehydrate tissues were clearly demonstrated to dissolve CCs in atherosclerotic plaques. In the late 1700s ethanol was introduced for tissue dehydration to trim specimens for light microscopy. A similar formulation of ethanol is currently used in tissue preparation for SEM as well. Although pathologists knew for a long time that CCs were dissolved and a residual tissue imprint or clefts representing empty spaces were recognized, the lumen of the vessel where many CCs were ejected was dissolved, and without a tissue imprint those CCs became invisible. Also, CCs perforating the intima were not appreciated but became visible by SEM with use of vacuum dehydration instead of ethanol dehydration followed by examination of the luminal surface.Thus the extent and the type of injury induced by CCs became recognized. To con firm that these observations were not related to the vacuum treatment, fresh plaques were examined by fluorescence microscopy without any tissue processing, and they also demonstrated findings similar to CCs perforating the intima (Figure 2). These data also have clinical implications since moderate ethanol intake by humans has demonstrated a beneficial effect on reducing the risk of ACS and strokes [35].
The association of CCs with ruptured plaque was further established in an autopsy study where a total of 215 coronary artery plaque disruptions were reported in 83 patients, with 77% of patients having multiple sites of plaque disruptions [36]. Parallel alignment of CCs within the necrotic core was present in 59% of all disrupted plaques regardless of plaque size or the severity of luminal stenosis. It was concluded that parallel con figuration of CCs may be a marker of plaques prone to rupture. Research efforts are under way to characterize plaque details with use of advanced imaging modalities to identify vulnerable plaques.
Inflammation in Response to Plaque Rupture/Injury
By application of the model of CC volume expansion on plaque injury, it becomes possible to recognize how both plaque rupture and plaque erosion are essentially a continuum of the same process. As CCs expand the LRNC, they begin to pierce their way through the soft tissues, including the fibrous cap [15, 17, 18] (Figure 4). A relatively small LRNC may not have a sufficient amount of cholesterol to produce enough CCs to induce sufficient expansion to rupture the fibrous cap; thus the CCs may perforate only the intimal surface. This can eventually lead to thrombus formation but that may not be as dramatic an event as with those plaques with a large necrotic core that have enough CC formations that expand rapidly to tear the fibrous cap. These findings could also help explain some of the clinical variances observed between men and women with regard to ACS presentation. Magnetic resonance studies have demonstrated that for the same degree of arterial lumen stenosis, women typically have smaller necrotic cores compared with their men counterparts [37]. They are also known to have more erosion than ruptures [38]. Thus their symptoms differ from and are less dramatic in presentation compared with those of men, who have larger necrotic cores and present more often with rupture and classic symptoms of crushing chest pain.Moreover, the perforation of the intima by CCs without an actual infarct could trigger a systemic inflammatory process. This leads to production of NLPR3 and subsequently C-reactive protein, which is a marker for increased risk of cardiovascular events [28]. The associated risk and prediction of worse clinical outcomes was first noted in patients with unstable angina by Liuzzo et al. [39].
Several crystalline substances that are either foreign or native to cells have been demonstrated to induce sterile inflammation via NLRP3 inflammasome activation. Silica crystals, monosodium urate crystals, and CCs induce NLRP3, which leads to the release of the active forms of the inflammatory cytokines IL-1β, IL-1α, and IL-18 via caspase 1[25]. In atherosclerotic lesions, CCs can be found in intracellular and extracellular spaces. Intracellular CCs may result from phagocytosis of extracellular CCs or may arise as a result of the excessive uptake of oxidized LDL by macrophages that is mediated by the scavenger receptor CD36 [40]. The free cholesterol released from oxidized LDL can lead to the formation of intracellular CCs within lysosomes that damage these lysosomes and activate NLRP3 and promote the release of IL-1β [41].
Extracellular CCs that form in atherosclerotic lesions can also induce the release of inflammatory cytokines through an alternative pathway.These CCs are recognized as a danger or damage signal via damage-associated molecular pattern receptors on cell membranes. In humans the receptor has been identified as macrophage-inducible C-type lectin (Mincle), which is expressed on activated macrophages and dendritic cells [42]. Mincle selectively binds crystalline cholesterol, the closely related plant sterol sitosterol, and the cholesterol synthesis intermediate desmosterol but does not bind to the sterol hormones progesterone, estradiol,testosterone, aldosterone, or dehydroepiandrosterone. On binding CCs, Mincle acts via the nuclear factor κβ pathway to induce the synthesis of pro-IL-1β and also likely activates NLRP3, leading to the subsequent cleavage of pro-IL-1β and release of activated IL-1β. Notably, human Mincle recognizes and binds to CCs, while the corresponding Mincle from mouse and rat does not bind to CCs, reflecting a possible evolutionary adaptation in humans to recognize CCs and activate innate immune responses.
What Occludes the Artery During Myocardial Infarction?
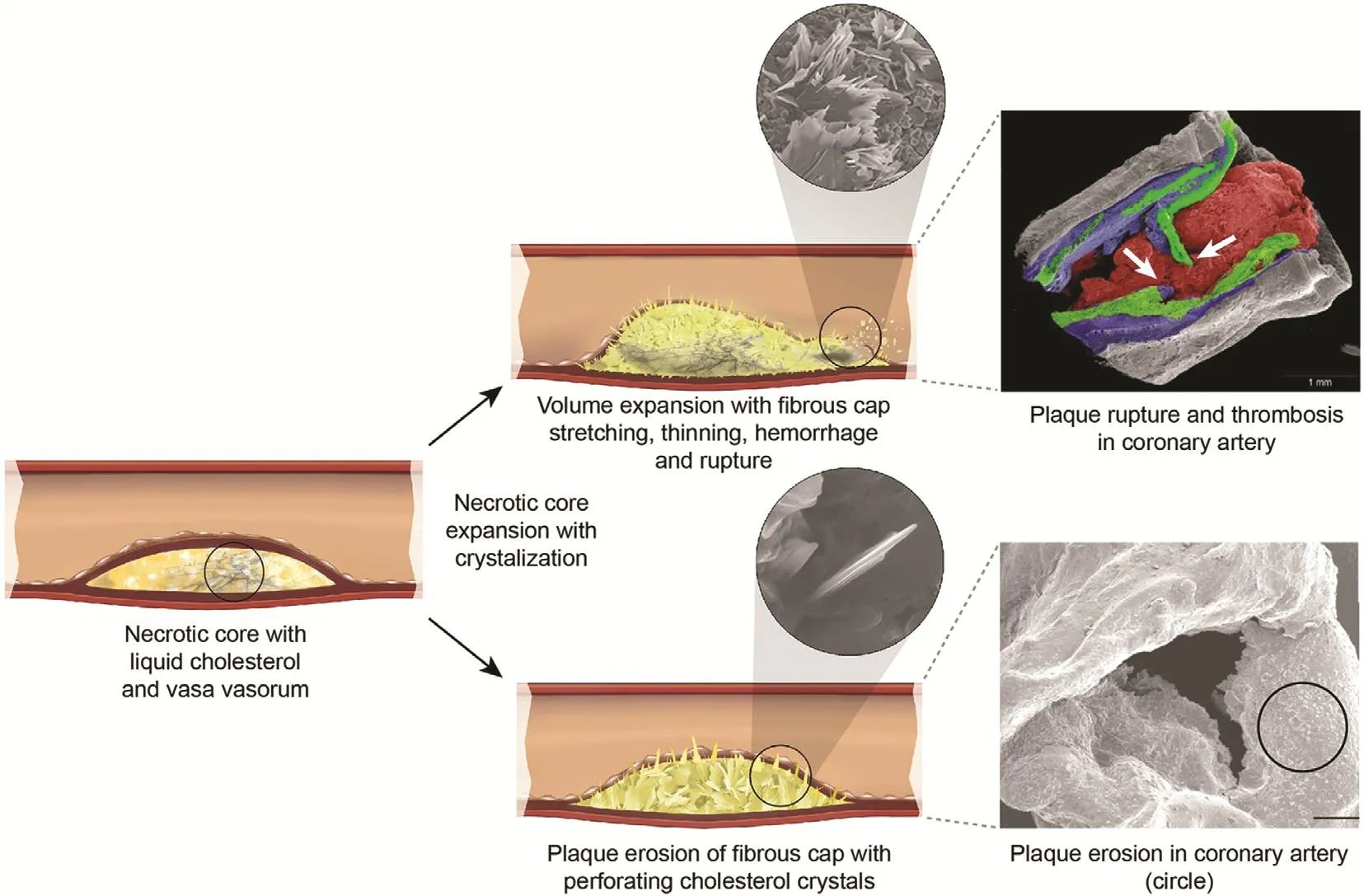
Figure 4 Mechanism of Plaque Hemorrhage, Rupture, and/or Erosion Induced by Cholesterol Crystallization with Volume Expansion of the Necrotic Core.
The actual event had been elusive for decades. In 1912 Herrick [43] proposed that thrombosis was the major culprit in AMI and cited the acute autopsy of a famous Danish music conductor that demonstrated a“thrombus” obstructing the coronary artery. However,subsequent autopsies of patients who died of myocardial infarction did not consistently demonstrate evidence of a thrombus, so it was considered that thrombus may be a postmortem event [44]. A possible explanation for these contradictory observations may be related to refrigeration. In the modern era, most autopsies are not performed immediately following death, but the bodies are usually kept refrigerated for several days, giving ample time for the thrombus to lyse. However, urgent coronary artery bypass surgery and angiography performed during AMI have reestablished the acute presence of thrombus during AMI[45], and the subsequent use of thrombolytic agents became popularized [46]. However, presently, arterial obstruction during AMI is usually treated mechanically with percutaneous intervention [47]. Yet, the story does not end here because many of the culprit arteries are not occluded by thrombus alone, and some arteries can be occluded predominantly or totally by CCs that were ejected into the lumen from a ruptured plaque, and thrombosis then becomes a secondary event [8, 9, 48] (Figure 5). Moreover, CCs traveling downstream from the ruptured site can injure the intimal surface, leading to vasospasm of the coronary artery further aggravating the injury and often causing a “no-re flow” phenomenon following percutaneous coronary intervention [49] (Figure 6). Furthermore,embolized CCs in the distal circulation can lodge in the myocardium and trigger an inflammatory injury to the myocardium in addition to ischemic injury [50].
Muscle Inflammation During Myocardial Infarction
Although ischemia is the major cause of myocardial injury and subsequent inflammation, CCs have been shown to cause muscle inflammation and injury independent of ischemia. This was demonstrated in a recent study where CC emboli without arterial obstruction were capable of inducing inflammation and muscle necrosis [50]. In humans, myocardial infarction without arterial occlusion has become recognized as an entity especially in female patients[51, 52]. With use of a rabbit model where a small amount of CCs were embolized in the distal femoral artery, inflammation and muscle injury was detected with patent arteries and normal perfusion by fluorodeoxy glucose on PET/CT. This is similar to what has been recognized in cholesterol embolic syndrome in the peripheral circulation [51]. However,extensive release of CCs into small arteries such as the coronary circulation can cause mechanical obstruction with ischemia and trigger direct muscle injury by inflammation. These findings are critical to the understanding of injury during ACS.
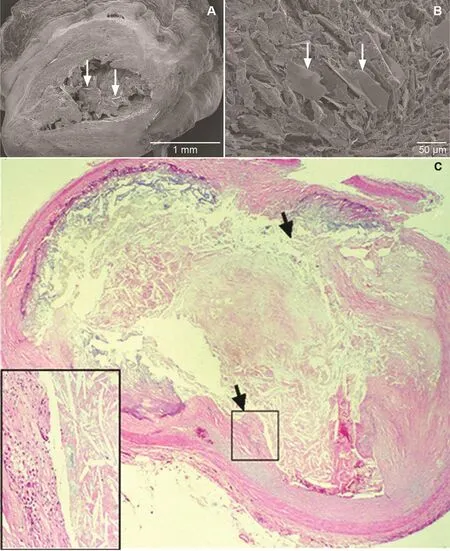
Figure 5 Scanning and Light Micrographs of Coronary Arteries of Patients Who Died of Acute Myocardial Infarction.
Stroke
The cause of stroke is multifactorial, but with advancing age ischemic strokes become increasingly common. Carotid artery stenosis (CAS) secondary to atherosclerosis has been attributed as an underlying mechanism in nearly 20% of all ischemic strokes [53]. The plaque morphology, including the fibrous cap, LRNC, and inflammatory cells, is similar to what is seen in the coronary arteries. The
location of the plaque is determined by many factors, including hydrodynamic factors such as shear wall stress. The carotid bulb is the commonest site for formation of the atherosclerotic plaque. Also,younger patients had higher levels of inflammatory cellular in filtrates and lower plaque calcification [54]. Moreover, embolization of CCs has been known to cause amaurosis fugax, and CCs are often seen lodged in the retinal arteries by fundoscopic examination [55]. Studies in rabbits and mice have demonstrated that CC emboli in the brain can cause inflammation with loss of cognitive function without necessarily causing an ischemic stroke [56, 57].However, when extensive amounts of CCs are used for embolization, micro infarcts can be detected[58]. This raises the implication of the role of CCs in neurovascular disease. Moreover, CCs have been detected in blood samples from patients who abuse alcohol. It was suggested that vascular inflammation is mediated by NLRP3/caspase 1 as the underlying mechanism for alcohol-medicated neuronal degeneration [59]. This adds to the growing body of evidence that CCs induce vascular inflammation and end organ injury.
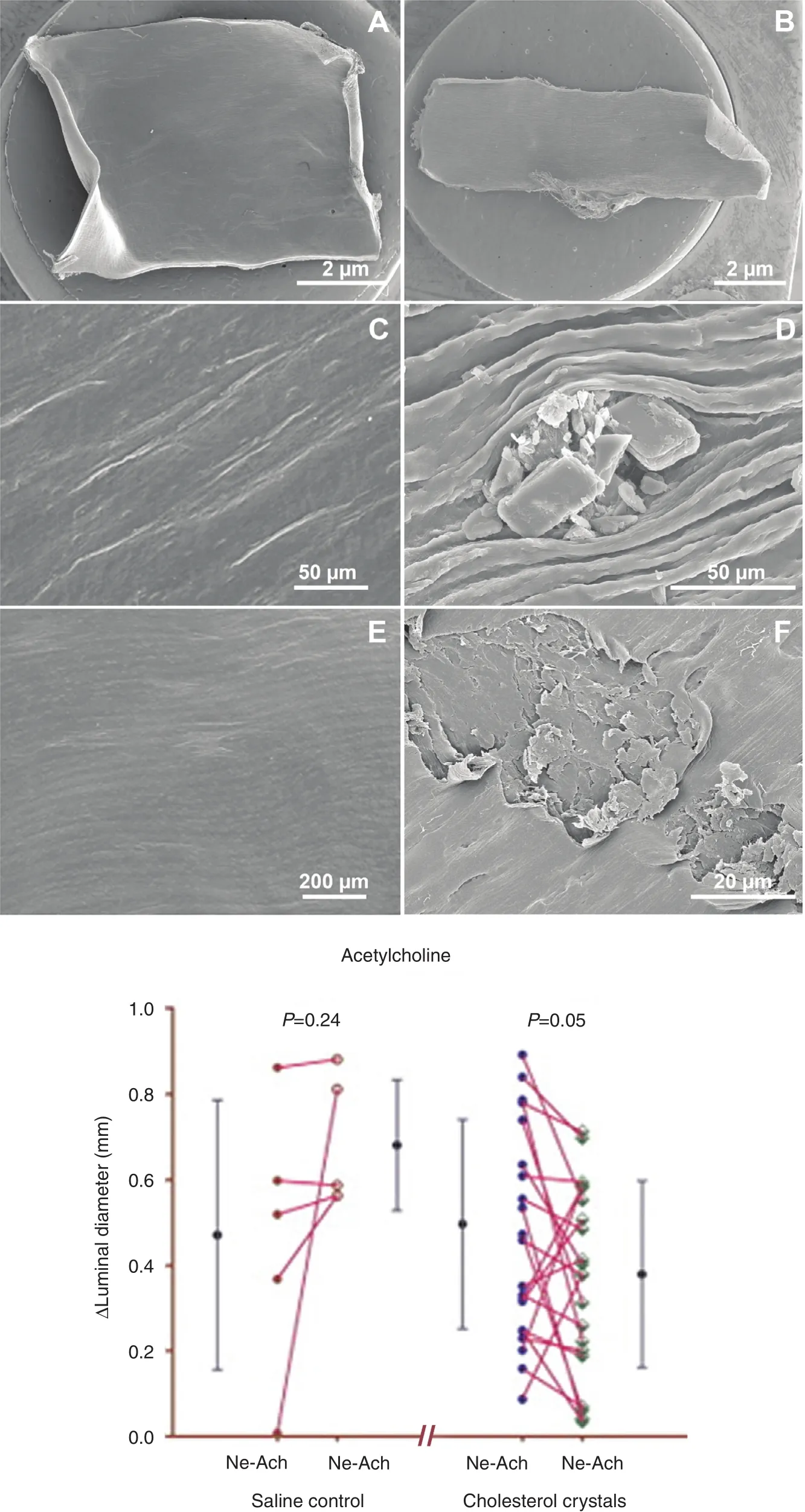
Figure 6 Scanning Electron Micrographs of Arterial Intima from Vessels Exposed to Circulating Saline With and Without Cholesterol Crystals.
Natural History of Carotid Artery Atherosclerosis
Atherosclerotic plaque in carotid arteries is believed to be present in up to 10% patients older than 65 years. On the basis of the absence or presence of clinical symptoms (transient ischemic attack or stroke), these patients are classified as having asymptomatic or symptomatic CAS respectively.
The relationship between carotid artery atherosclerosis, its clinical manifestations in the form of transient ischemic attack and stroke, and their response to medical or surgical treatment has been extensively studied in several population studies, such as the North American Symptomatic Carotid Endarterectomy Trial (NASCET), the Asymptomatic Carotid Surgery Trial (ACST), and the Asymptomatic Carotid Atherosclerotic Study(ACAS) [60–62].
NASCET reported a strong clinical correlation between the risk of stroke and the severity of CAS,a relationship that is ambiguous in asymptomatic patients. Both ACAS and ACST reported improved clinical outcomes associated with carotid endarterectomy as compared with medical therapy alone in asymptomatic CAS patients. However, medical therapy has evolved significantly since the completion of these trials, and could be responsible for lack of superiority in clinical outcomes of surgery compared with contemporary medical therapy with aspirin and statins in ACAS data since 2001 [63].
Imaging studies assist in determining the luminal stenosis and also in characterizing the plaque morphology, which improve our understanding of the natural history of the atherosclerotic plaque.The accessibility of the carotid arteries to a variety of noninvasive imaging techniques (i.e., Doppler ultrasonography, PET, MRI) allows detailed resolution of atherosclerotic plaque morphology. For example, high-resolution MRI can provide details of the carotid plaque such as plaque hemorrhage,LRNC size, and fibrous cap thickness. These findings have been shown to be well correlated with histologic findings [64, 65] (Figure 7). Animal and human studies using PET/CT scanning have shown the ability to detect vulnerable plaque especially in carotid plaques [66, 67]. The current management of CAS is based on the symptoms, mainly stroke/transient ischemic attack. A surgical and percutaneous approach can be used depending the anatomy and comorbidities.
Potential Therapeutic Approaches
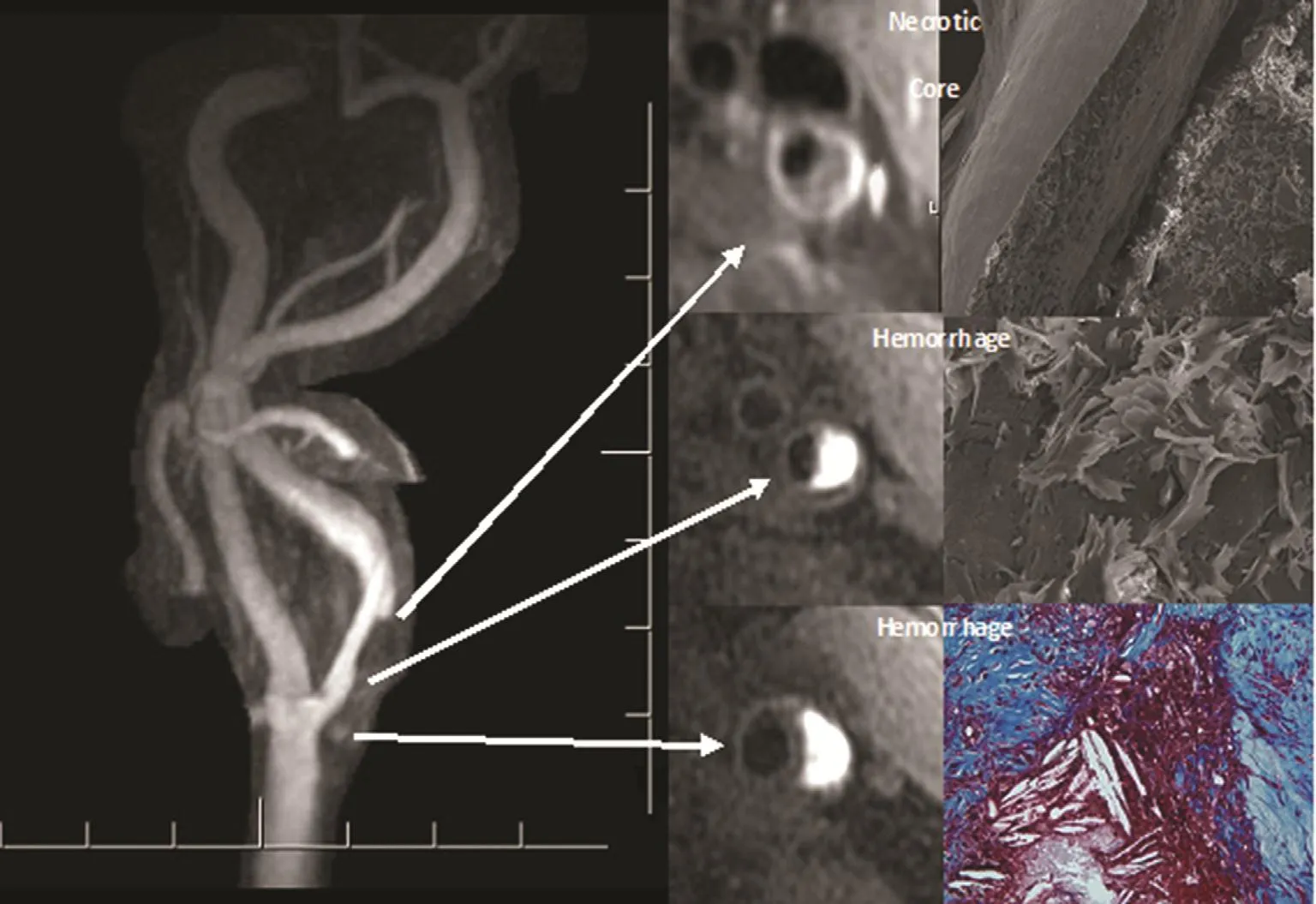
Figure 7 Left Carotid Artery on 500-μm Resolution Contrast-Enhanced Magnetic Resonance Angiogram.
Statins, aspirin, and ethanol are known to dissolve CCs, and may in part exert their clinical benefits in prevention of ACS and CVAs via this mechanism[17–19, 68]. Some of the potential pleotropic effects of statins are recognized to be the reduction in vascular inflammation and plaque stabilization [69].Moreover, statins have also been shown to directly dissolve CCs, which could contribute to reduction of inflammation and subsequent plaque instability [17,48] (Figure 8). Also, lowering of cholesterol levels by other approaches was demonstrated in an atherosclerotic rabbit model where ezetimibe was used to reduce the absorption of cholesterol. This resulted in reduced arterial wall CC content, serum inflammatory biomarker response, and plaque rupture and thrombosis [70, 71]. Corroboratory findings were reported in a recent clinical trial using ezetimibe,the Improved Reduction of Outcomes Vytorin Efficacy International Trial (Improve-IT), which also demonstrated the potential benefits of additional LDL level lowering [72]. Other approaches have used cyclodextrin conjugated with superparamagnetic iron oxide nanoparticles for selective binding and detection of CCs as well as a therapeutic treatment to regress atherosclerosis [73, 74].Several studies are under way that propose blocking the inflammatory process so as to stabilize, prevent,or even regress atherosclerotic plaques and lower the incidence of ACS events. These include the Anti-inflammatory Thrombosis Outcomes Study(CANTOS) using canakinumab, which is an IL-1β inhibitor [75], the Cardiovascular Inflammation Reduction Trial (CIRT) with methotrexate, a dihydrofolate reductase inhibitor that inhibits DNA synthesis [76], use of colchicine, a mitosis inhibitor that also blocks neutrophil motility, reducing inflammation [77], and use of antileukotriene agents, lipoxins and resolvins, which are being investigated for their anti-inflammatory effects [78].
Other Inflammatory Causes of Plaque Instability
Several other causes of plaque instability are under investigation. These include the potential role of gut bacteria mediated by trimethylamineN-oxide, a gut metabolite of dietary animal proteins that is associated with coronary artery disease [79]. Also, bacteria have been shown to be attracted to CCs as a potential nutrient source [80]. All these bacterial metabolic activities could result in plaque instability. Also, other immunemediated responses involving heat shock proteins have been investigated. Heat shock protein 60 was shown to be expressed in coronary plaques with an associated rise in C-reactive protein levels [81]. Research in this field is ongoing and may be relevant to plaque rupture.
Summary
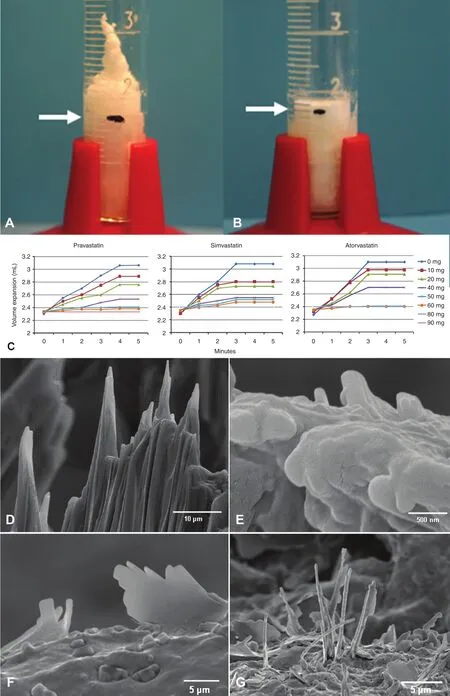
Figure 8 Effect of Statins on Cholesterol Crystallization and Crystal Morphology.
Cholesterol accumulated in the arterial wall exerts its effects by crystallizing and inducing both a local and a systemic inflammatory response as well as causing mechanical injury to the soft tissues of the arterial wall to rupture the plaque. This process occurs because cholesterol expands in volume during crystallization. Release of CCs into the circulation can obstruct small vessels such as coronary arteries or erode the surface and cause thrombosis.Also, as they travel downstream, CCs can scrape the intima and induce vasospastic activity. Furthermore,CCs lodged in the muscle can trigger an inflammatory reaction that can result in muscle cell death. All these effects of CCs can occur acutely or chronically, which may account for variation in clinical presentation. Therapeutic approaches would include use of agents that inhibit CC formation or dissolve CCs. Other approaches could include inhibition of the associated inflammation to reduce the extent of vascular and end organ injury. Certainly, statins and aspirin seem to have those properties to fulfil this task. Also, moderate alcohol consumption has been shown to dissolve CCs and reduce the risk of ACS and stroke. Other agents that are safer to use could be developed with similar pharmacologic effects.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
1. World Health Organization. The top 10 causes of death. 2017 http://www.who.int/mediacentre/factsheets/fs310/en/.
2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics–2016 update: a report from the American Heart Association.Circulation 2016;133(4): e38–360.
3. Constantinides P. Plaque fissures in human coronary thrombosis. J Atheroscler Res 1966;6:1–17.
4. Davies MJ, Thomas AC. Plaque fissuring: the cause of acute myocardial infarction causing sudden ischaemic death, and crescendo angina. Br Heart J 1985;53:363–73.
5. Barger AC, Beeuwkes R 3rd,Lainey LL, Silverman KJ.Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis.N Engl J Med 1984;310:175–7.
6. Maseri A, Davies G, Hackett D, Kaski JC. Coronary artery spasm and vasoconstriction. The case for a distinction. Circulation 1990;81:1983–91.
7. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 1994;94:2494–503.
8. Abela, GS, Aziz, K. Cholesterol crystals rupture biological membranes and human plaques during acute cardiovascular events: a novel insight into plaque rupture by scanning electron microscopy.Scanning 2006;28:1–10.
9. Abela, GS, Aziz, K. Cholesterol crystals cause mechanical damage to biological membranes: a proposed mechanism of plaque rupture and erosion leading to arterial thrombosis. Clin Cardiol 2005;28:413–20.
10. Schwenke DC, Carew TE. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis 1989;9:908–18.
11. Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987;316:1371–5.
12. Muller JE, Abela GS, Nesto RW,To fler GH. Triggers, acute risk factors, and vulnerable plaques: the lexicon of a new frontier. J Am Coll Cardiol 1994;23:809–13.
13. Schaar JA, Muller JE, Falk E,Virmani R, Fuster V, Serruys PW,et al. Terminology for high-risk and vulnerable coronary artery plaques.Report of a meeting on the vulnerable plaque, June 17 and 18, 2003,Santorini, Greece. Eur Heart J 2004;25:1077–82.
14. Stone GW, Maehara A, Lansky AJ,de Bruyne B, Cristea E, Mintz GS,et al. A prospective natural-history study of coronary atherosclerosis.N Engl J Med 2011;364:226–35.
15. Abela GS. Cholesterol crystals piercing the arterial plaque and intima triggers local and systemic inflammation. J Clin Lipidol 2010;4:156–64.
16. Kellner-Weibel G, Yancy PG, Jerome WG, Walser T,Mason RP, Phillips MC, et al.Crystallization of free cholesterol in model macrophage foam cells.Arterioscler Thromb Vasc Biol 1999;19:1991–8.
17. Abela GS, Vedre A, Janoudi A,Huang R, Durga S, Tamhane U.Effect of statins on cholesterol crystallization and atherosclerotic plaque stabilization. Am J Cardiol 2011;107:1710–7.
18. Abela GS, Aziz K, Vedre A, Pathak D, Talbott JD, DeJong J. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am J Cardiol 2009;103:959–68.
19. Nasiri M, Janoudi A, Vanderberg A, Frame M, Flegler C, Flegler S,et al. Role of cholesterol crystals in atherosclerosis is unmasked by altering tissue preparation methods.Micros Res Tech 2015;78:969–74.
20. Liu L, Gardecki JA, Nadkarni SK, Toussaint JD, Yagi Y, Bouma BE, et al. Imaging the subcellular structure of human coronary atherosclerosis using microoptical coherence tomography. Nat Med 2011;17:1010–4.
21. Tian J, Ren X, Vergallo R, Xing L, Yu H, Jia H, et al. Distinct morphological features of ruptured culprit plaque for acute coronary events compared to those with silent rupture and thin-cap fibroatheroma: a combined optical coherence tomography and intravascular ultrasound study. J Am Coll Cardiol 2014;63:2209–16.
22. Nakamura S, Inami S, Murai K,Takano M, Takano H, Asai K,et al. Relationship between cholesterol crystals and culprit lesion characteristics in patients with stable coronary artery disease:an optical coherence tomography study. Clin Res Cardiol 2014;103:1015–21.
23. Vedre A, Pathak DR, Crimp M,Lum C, Koochesfahani M, Abela GS. Physical factors that trigger cholesterol crystallization leading to plaque rupture. Atherosclerosis 2009;203:89–96.
24. Abela GS. Plaque rupture by cholesterol crystallization. Cardiosource(American College of Cardiology,Atlanta, GA, March 13, 2006)ACCEL;8;2006, CD #2; #2.
25. Düwell P, Kono H, Rayner KJ, Sirois CM, Vladimer G,Bauernfeind F, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease.Nature 2010;464:1357–62.
26. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S,Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages:a novel link between cholesterol metabolism and inflammation.PLoS One 2010;5:e11765. doi:10.1371/journal.pone.0011765.
27. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J.Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237–41.
28. Ridker PM. From CRP to IL-6 to IL-1: moving upstream to identify novel targets for atheroprotection.Circ Res 2016;118:145–56.
29. Loree HM, Kamm RD,Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res 1992;71:850–8.
30. Lundberg B. Chemical composition and physical state of lipid deposits in atherosclerosis. Atherosclerosis 1985;56:93–110.
31. Muller JE, To fler GH, Stone PH.Circadian variation and triggers of onset of acute cardiovascular disease. Circulation 1989;79:733–43.
32. Fares A. Winter cardiovascular diseases phenomenon. N Am J Med Sci 2013;5:266–79.
33. Franklin BA, George P, Henry R,Gordon S, Timmis GC, O’Neill WW. Acute myocardial infarction after manual or automated snow removal. Am J Cardiol 2001;87:1282–3.
34. Curran RC. Colour atlas of histopathology. 3rd ed. New York: Oxford University, Harvey Miller; 1985.p. 103.
35. Fernández Jarne E, Martínez Losa E, Serrano Martínez M,Prado Santamaría M, Brugarolas Brufau C, Martínez González MA. Type of alcoholic beverage and first acute myocardial infarction: a case control study in a Mediterranean country. Clin Cardiol 2003;26:313–8.
36. Frink RJ. Parallel cholesterol crystals: a sign of impending plaque rupture? J Invasive Cardiol 2010;22:406–11.
37. Ota H, Reeves MJ, Zhu DC,Majid A, Collar A, Yuan C, et al.Sex differences in patients with asymptomatic carotid atherosclerotic plaque: in vivo 3.0-T magnetic resonance study. Stroke 2010;41:1630–5.
38. Arbustini E, Dal Bello B, Morbini P,Burke AP, Bocciarelli M, Specchia G, et al. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction.Heart 1999;82:269–72.
39. Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG,Pepys MB, et al. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med 1994;331:417–24.
40. Kunjathoor VV, Febbraio M,Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 2002;277:49982–8.
41. Sheedy FJ, Grebe A, Rayner KJ,Kalantari P, Ramkhelawon B,Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 201314(8):812–20. doi: 10.1038/ni.2639.
42. Kiyotake R, Oh-Hora M, Ishikawa E, Miyamoto T, Ishibashi T,Yamasaki S. Human Mincle binds to cholesterol crystals and triggers innate immune responses. J Biol Chem 2015;290:25322–32.
43. Herrick JB. Clinical features of sudden obstruction of the coronary arteries. J Am Med Assoc 1912;59:2015–20.
44. Roberts WC, Buja LM. The frequency and significance of coronary arterial thrombi and other observations in fatal acute myocardial infarction: a study of 107 necropsy patients. Am J Med 1972;52:425-43.
45. DeWood MA, Spores J, Notske R, Mouser LT, Burroughs R,Golden MS, et al. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med 1980;303:897–902.
46. Rentrop KP, Blanke H, Karsch KR,Wiegand V, Köstering H, Oster H,et al. Acute myocardial infarction:intracoronary application of nitroglycerin and streptokinase. Clin Cardiol 1979;2:354–63.
47. The Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes(GUSTO IIb) Angioplasty Substudy Investigators. A clinical trial comparing primary coronary angioplasty with tissue plasminogen activator for acute myocardial infarction. N Engl J Med 1997;336:1621–8.
48. Janoudi A, Shamoun FE,Kalavakunta JK, Abela GS.Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J 2016;37:1959–67.
49. Gadeela N, Rubinstein J,Tamhane U, Huang R, Pathak DR, Hosein H-A, et al. The impact of circulating cholesterol crystals on vasomotor function: implications for no-reflow phenomenon J Am Coll Cardiol Interv 2011;4:521–9.
50. Pervaiz MH, Durga S, Janoudi A,Berger K, Abela GS. PET/CTA detection of muscle inflammation related to cholesterol crystal emboli without arterial obstruction.J Nucl Cardiol 2017. doi:10.1007/s12350-017-0826-y.
51. Ghanem F, Vodnala D, Kalavakunta JK, Durga S, Thormeier N,Subramaniyan P, et al. Cholesterol crystal embolization following plaque rupture: a systemic disease with unusual features. J Biomed Res 2017;31:1–13.
52. Niccoli G, Scalone G, Crea F.Acute myocardial infarction with no obstructive coronary atherosclerosis: mechanisms and management. Eur Heart J 2015;36:475–81.
53. Petty GW, Brown RD Jr, Whisnant JP, Sicks JD, O’Fallon WM,Wiebers DO. Ischemic stroke subtypes: a population-based study of incidence and risk factors. Stroke 1999;30:2513–6.
54. Redgrave JN, Lovett JK, Rothwell PM. Histological features of symptomatic carotid plaques in relation to age and smoking: the oxford plaque study. Stroke 2010;41:2288–94.
55. Hollenhorst RW. Significance of bright plaques in the retinal arterioles. J Am Med Assoc 1961;178:23–9.
56. Rapp JH, Pan XM, Neumann M,Hong M, Hollenbeck K, Liu J.Microemboli composed of cholesterol crystals disrupt the bloodbrain barrier and reduce cognition.Stroke 2008;39:2354–61.
57. Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K,Liu C, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale.Ann Neurol 2010;67:221–9.
58. Wang M, Iliff JJ, Liao Y, Chen MJ,Shinseki MS, Venkataraman A,et al. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J Neurosci 2012;32:17948–60.
59. Abdul-Muneer PM, Alikunju S,Mishra V, Schuetz H, Szlachetka AM, Burnham EL, et al. Activation of NLRP3 inflammasome by cholesterol crystals in alcohol consumption induces atherosclerotic lesions. Brain Behav Immun 2017;62:291–305.
60. Ferguson GG, Eliasziw M, Barr HW, Clagett GP, Barnes RW,Wallace MC, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke 1999;30:1751–8.
61. Halliday A, Harrison M, Hayter E, Kong X, Mans field A, Marro J,et al. 10-year stroke prevention after successful carotid endarterectomy for asymptomatic stenosis (ACST-1): a multicentre randomised trial.Lancet 2010;376:1074–84.
62. Walker MD, Marler JR, Goldstein M, Grady PA, Toole JF, Baker WH,et al. Endarterectomy for asymptomatic carotid artery stenosis. J Am Med Assoc 1995;273:1421–8.
63. Abbott AL. Medical (nonsurgical) intervention alone is now best for prevention of stroke associated with asymptomatic severe carotid stenosis: results of a systematic review and analysis. Stroke 2009;40(10):e573–83.
64. Cai J, Hatsukami TS, Ferguson MS, Kerwin WS, Saam T, Chu B,et al. In vivo quantitative measurement of intact fibrous cap and lipid-rich necrotic core size in atherosclerotic carotid plaque:comparison of high-resolution,contrast-enhanced magnetic resonance imaging and histology.Circulation 2005;112:3437–44.
65. Mughal MM, Khan MM, DeMarco K, Majid A, Shamoun F, Abela GS.Symptomatic and asymptomatic carotid artery plaque. Expert Rev Cardiovasc Ther 2011;9:1315–30.www.tandfonline.com.
66. Aziz K, Berger K, Claycombe K,Huang R, Patel R, Abela GS. Noninvasive detection and localization of vulnerable plaque and arterial thrombosis using CTA/PET.Circulation 2008;117:2061–70.
67. Tawakol A, Migrino RQ, Hoffmann U, Abbara S, Houser S, Gewirtz H,et al. Noninvasive in vivo measurement of vascular inflammation with F-18 fluorodeoxyglucose positron emission tomography. J Nucl Cardiol 2005;12:294–301.
68. Vedre A, Aziz K, Huang R, Abela GS. Aspirin prevents cholesterol crystallization: a potential mechanism of plaque stabilization. J Am Coll Cardiol 2008;51(Suppl A):318.
69. Crisby M, Nordin-Fredriksson G,Shah PK, Yano J, Zhu J, Nilsson J.Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation 2001;103:926–33.
70. Patel R, Janoudi A, Vedre A, Aziz K, Tamhane U, Rubinstein J, et al.Plaque rupture and thrombosis is reduced by lowering cholesterol levels and crystallization with ezetimibe and is correlated with FDG-PET. Arterioscler Thromb Vasc Biol 2011;31:2007–14.
71. Gómez-Garre D1, Muñoz-Pacheco P, González-Rubio ML, Aragoncillo P, Granados R, Fernández-Cruz A.Ezetimibe reduces plaque inflammation in a rabbit model of atherosclerosis and inhibits monocyte migration in addition to its lipid-lowering effect.Br J Pharmacol 2009;156:1218–27.
72. Cannon CP, Blazing MA,Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015;372:2387–97.
73. Li H, El-Dakdouki MH, Zhu DC,Abela GS, Huang X. Synthesis of β-cyclodextrin conjugated superparamagnetic iron oxide nanoparticles for selective binding and detection of cholesterol crystals.Chem Commun 2012;48:3385–7.
74. Zimmer S, Grebe A, Bakke SS,Bode N, Halvorsen B, Ulas T, et al.Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med 2016;8(333):333ra50. doi:10.1126/scitranslmed.aad6100.
75. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 2011;162:597–605.
76. Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J,Zaharris E, et al. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J 2013;166:199–207.
77. Nidorf SM, Eikelboom JW,Budgeon CA, Thompson PL. Lowdose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 2013;61:404–10.
78. Hersberger M. Potential role of the lipoxygenase derived lipid mediators in atherosclerosis: leukotrienes, lipoxins and resolvins. Clin Chem Lab Med 2010;48:1063–73.
79. Tang WH, Wang Z, Kennedy DJ,Wu Y, Buffa JA, Agatisa-Boyle B,et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO)pathway contributes to both development of renal insuf ficiency and mortality risk in chronic kidney disease. Circ Res 2015;116:448–55.
80. Baig I, Kalra A, Scharmen A, Raju M, Gardiner J, De Feijter-Rupp H, et al. Atherosclerotic arteries with cholesterol crystals enhance bacterial growth: risk for plaque destabilization. J Clin Lipidol 2016;10:A177.
81. Andrié RP, Bauriedel G, Braun P,Höpp HW, Nickenig G, Skowasch D. Prevalence of intimal heat shock protein 60 homologues in unstable angina and correlation with anti-heat shock protein antibody titers. Basic Res Cardiol 2011;106:657–65.
 Cardiovascular Innovations and Applications2017年2期
Cardiovascular Innovations and Applications2017年2期
- Cardiovascular Innovations and Applications的其它文章
- Relationship Between Morning Hypertension and T-Peak to T-End Interval in Patients with Suspected Coronary Heart Disease
- Occlusive Spasm of the Left Anterior Descending Artery and First Diagonal Branch After Implantation of Everolimus Eluting Stents Without Re-stenosis in a Female Patient with Resting Angina
- Chronic Kidney Disease is a New Target of Cardiac Rehabilitation
- Evaluation of Multidisciplinary Collaborative Care in Patients with Acute Coronary Syndrome and Depression and/or Anxiety Disorders
- Digoxin and Heart Failure: Are We Clear Yet?
- Telemedicine: Its Importance in Cardiology Practice. Experience in Chile