
Comparison of chicoric acid,and its metabolites caffeic acid and caftaric acid:In vitro protection of biological macromolecules and inflammator responses in BV2 microglial cells
2017-05-22 12:43:54QianLiuFanLiuLilingZhangYajieNiuZhigangLiuXueboLiu
食品科学与人类健康(英文) 2017年4期
Qian Liu,Fan Liu,Liling Zhang,Yajie Niu,Zhigang Liu,Xuebo Liu
Laboratory of Functional Chemistry and Nutrition of Food,College of Food Science and Engineering,Northwest A&F University,Yangling,China
Abstract Chicoric acid (CA),a natural phenolic acid,has been used as a nutraceutical food ingredient due to its powerful antioxidant,anti-HIV and anti-diabetic bioactivities.CA could be partly metabolized into caffeic acid (CFA) and caftaric acid (CTA) on cytochrome P450s in rat liver microsomes.To compare the protective effects of CA and its metabolites on biomolecules and inflammator responses,oxidative damage induced by free radicals in vitro and microglial inflammatio triggered by lipopolysaccharide in BV2 cells were constructed.Results showed that CA,CTA and CFA all significantl inhibited protein degradation and carbonylation induced by hydroxyl radicals and alcoxyl radicals,and suppressed hemin/nitrite/H2O2 triggered-nitration.Moreover,CA,CTA and CFA all exerted remarkable inhibition capacities on linoleic acid and soybean lecithin liposomes peroxidation in a dose-dependent manner,and restrained the oxidation of herring sperm DNA,as well as the breakage of pBR322 plasmid DNA.Furthermore,CA and its metabolites suppressed lipopolysaccharide-induced decline of BV2 cell viability and the production of NO and ROS.However,bioactivities of CA were significantl stronger than those ofits metabolites within a certain concentration range.This study provides scientifibasis for the application of CA and its metabolites as nutrition and natural antioxidants.
Keywords: Chicoric acid and its metabolites;Oxidative damage;Biomolecules;Microglia;Inflammatio
1.Introduction
sive ROS/RNS will attack biological macromolecules such as proteins,lipids and DNA,disrupt cell structure and function,and also cause inflammatio responses [3,4].Nicotinamide adenine dinucleotide phosphate oxidase is a major source of ROS production in many non-phagocytic cells.Microglial cells produce inflammator cytokines such as TNF-α induced by activated nicotinamide adenine dinucleotide phosphate oxidase[5].ROS/RNS can also act as a second messenger to activate NFκB signaling pathway in microglia cells,causing gene expression related to inflammatio and lead to the production ofinflamma tory mediators,including TNF-α,IL-1β,NO and PGE2[6].
Phytochemical antioxidants have been widely used due to the higher natural and secure properties compared with synthetic antioxidants.A large number of studies have confirme that phytochemicals,especially plant phenols could prevent or alleviate oxidative stress.It has been reported that polyphenolic extract ofGeoffroea decorticans(cha~nar)exhibited antioxidant activities by scavenging ABTS+·,·OH,hydrogen peroxide,and protecting protein and lipid against oxidative damage [7].
Oxidative stress plays a pivotal role in the pathophysiology of diseases including neurodegenerative diseases,aging cancer,obesityandcardiovasculardiseases[1,2].Oncebeinginastateof oxidative stress or disease,generation of reactive oxygen species(ROS)/reactive nitrogen species(RNS)would increase.Exces-It has been proved that caffeic acid could improve cognitive function through inhibition oflipid peroxidation and NO production[8].Another study demonstrated that isoorientin could inhibit inflammator responses induced by lipopolysaccharide through ROS-related MAPK/NFκB signaling pathway in BV2 microglial cells[9].
Chicoric acid (CA),isolated from chicory [10],is widely distributed in the nature,which was also found in iceberg lettuce,dandelion,ocimumbasilicum,cymodoceanodosaandother plants[11,12].As a natural phenolic acid,CA has intense effects in anti-oxidation,anti-obesity,antiviral,and hpyerglycemic activities [13-15].Our previous report demonstrated that CA wasmetabolizedtocaffeicacid(CFA)andcaftaricacid(CTA)on cytochrome P450s in rat liver microsomes.CA and its metabolites all could significantl scavenge DPPH·,·OH,ABTS+·free radicals and had strong ferric reducing capacities in a dosedependent manner,among which,CA has the highest functional activities[16].However,comparisons on the protection abilities of biological macromolecules and anti-inflammator activities among CA and its metabolites have rarely been reported.
Therefore,this study is aimed to compare the inhibition of CA and its metabolites on the protein oxidation,carbonylation and nitration,lipid peroxidation and DNA oxidation breakage induced by hydroxyl radicals or alcoxyl radicals.Besides,in order to evaluate the effects of CA and its metabolites on lipopolysaccharide induced inflammatio in BV2 microglial cells,cell viability and the production of NO,ROS were detected.This study will improve our understanding of the antioxidant and anti-inflammator activities of CA and its metabolites,and provide a scientifibasis for the application of CA and its metabolites as nutrition and natural antioxidants.
2.Materials and methods
2.1.Chemicals and reagents
Chicoric acid,caffeic acid,and caftaric acid(purity ≥98%)were purchased from Weikeqi Biological Technology Co.,Ltd.(Sichuan,China).Bovine serum albumin (BSA),2,2-azobis (2-amidinopropane) dihydrochloride (AAPH),herring sperm DNA (hsDNA,D3159),linoleic acid (LA),Anti-DNP antibody,lipopolysaccharide,and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) were purchased from Sigma Chemical Co.(St.Louis,MO,USA).Soybean lecithin was obtained from Huamaike Biotechnology.,Ltd.(Beijing,China).PBR322 plasmid was purchased from Takara Shuzo Co.,Ltd.(Kyoto,Japan).Hydrogen peroxide (H2O2)was obtained from Sinopharm Group Chemical Reagent Co.,Ltd.(Shanghai,China).The BCA protein assay kit was obtained from Thermo Scientifi(Rockford,IL,USA).All other reagents were made in China and were of HPLC-grade or the highest commercially available grade.
2.2.BSA oxidative degradation
Hydroxyl free radicals generated by Cu2+react with H2O2will cause protein oxidation [17].The inhibition of CA and its metabolites on BSA oxidative degradation induced by Cu2+/H2O2were determined according to the previous method with some modifications CA,CFA,and CTA(10,50,100,500,1000 μmol/L)were mixed with 0.8 mg/mL BSA solution.H2O2(25 mmol/L) and CuSO4(0.1 mmol/L) were added to induce protein oxidative degradation followed by the pre-incubation at 37°C for 30 min.And the control group used pH 7.4 phosphate buffer instead of CuSO4and H2O2.After incubation at 37°C for 90 min,the protein content were measured by commasie brilliant blue staining,and the optical densities were analyzed by Quantity One 4.6.2.
Assays for the inhibition of CA and its metabolites on BSA oxidative degradation induced by AAPH were measured by the method described by Mayo et al.[18].The treatments were the similar with Cu2+/H2O2reaction system.Incubation at 37°C for 2 min made AAPH (50 mmol/L) thermal decomposition,producing alcoxyl radicals.After incubation at 37°C for 4 h,the protein content were measured by commasie brilliant blue staining,and the optical densities were analyzed by Quantity One 4.6.2.
2.3.BSA carbonylation
DNPH method was used to detect the degree of BSA carbonylation[19].400 μL samples were added into 4 mL DNPH solution,and placed the solution in the dark at room temperature for 1 h.Then mixed with 4 mL 10%icy trichloroacetic acid,and centrifugated at 4000 rmp at 4°C for 10 min,followed by washing precipitate three times with ethanol and ethyl acetate in 1:1 proportions.The absorbance of the precipitate dissolved in 4 mL guanidine hydrochloride was measured at 370 nm using a UV-vis spectrophotometer.The carbonyl contents were calculated using formula(1):
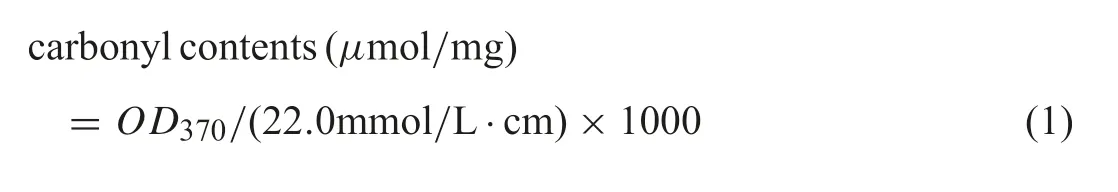
2.4.BSA nitration
Hemin/NaNO2/H2O2reaction system was used to induce protein nitration [20].CA,CFA,and CTA (10,50,100,500,1000 μmol/L) were mixed with 1.0 mg/mL BSA solution and preincubating for 5 min.And then hemin(100 μmol/L),NaNO2(10 mmol/L) and H2O2(1 mmol/L) were added.After incubation at 37°C for 30 min,the levels of nitration were measured by western blotting,and the optical densities were quantifie by Quantity One 4.6.2.
2.5.Western blotting
After the reaction,add 25 μL 5×SDS loading buffer to 100 μL reaction solution,and then immediately heated at 95°C for 10 min.The proteins were separated by SDS-PAGE and transferredontoPVDFmembranes(0.45 μm,Millipore).Blocking was carried out for 1.5 h in 5% nonfat dry milk in TBST buffer (20 mmol/L Tris,166 mmol/L NaCl,and 0.05% Tween 20,pH 7.5).Then,the membrane was washed 3 times each for 5 minusingTBSTbuffer.Theprimaryantibody3-NT(sc-32731,Santa Cruz,1:300)was added overnight at 4C.Then,after three washes in TBST,secondary antibodies (sc-2005,Santa Cruz)were added for 1.5 h.After another three washes with TBST,the immunoreactive bands were visualized with an enhanced chemiluminescence reagent(Thermo Fisher,China).
2.6.Lipidosome peroxidation
Lipidosome peroxidation was determined by previous methods [21].Mix CA,CFA,and CTA (10,50,100,500,1000 μmol/L)with 20 μL lipidosome(20 mg lecithin dissolved in 5 mL 0.05 mol/L pH 7.4 phosphate buffer fille with nitrogen)and preincubate for 10 min.Then 0.8 mmol/L FeSO4was added to induce peroxidation.After 37°C for 40 min,the reaction fin ished and the amount of thiobarbituric acid reactive substances(TBARS) induced by lipid peroxidation was observed by colorimetry.
2.7.Linoleic acid peroxidation
Fe2+/Vc reaction system was adopted to induce linoleic acid peroxidation [22].Linoleic acid (1 mmol/L) was mixed with CA,CFA,and CTA(10,50,100,500,1000 μmol/L).Reaction solution was vortexed with 50 μmol/L FeSO4and 1 mmol/L Vc,follow by 37°C water bath for 24 h away from light.The degree oflipid peroxidation was assessed by TBARS.
2.8.TBARS method
TBA method was used to measure the lipid peroxidation level[23].Reacting 10%trichloroacetic acid 100 μL and 0.8%thiobarbituric acid 100 μL with the samples in 100°C for 15 min.The reaction mixture was centrifuged for 10 min at 5000 r/min and the absorbance of the supernatant was measured at 532 nm using a UV-vis spectrophotometer.The inhibition of CA and its metabolites on lipid peroxidation induced by free radicals were calculated according to formula(2):
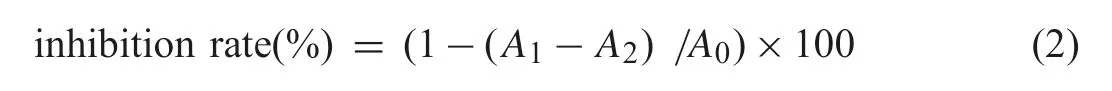
whereA1is the absorbance of the experimental group,A2is the absorbance of the induced group(using phosphate buffer instead of FeSO4solution),andA0is the absorbance of the control group(using phosphate buffer instead of the sample).
2.9.hsDNA oxidative damage
CA,CFA,and CTA (10,50,100,500,1000 μmol/L) were mixed with 2.0 mg/mL hsDNA.40 mmol/L AAPH was added to induce DNA oxidative damage.After incubation at 37°C for 12 h,the degree of DNA oxidative damage was measured by TBA method.
2.10.pBR322 plasmid DNA oxidative cleavage
By adopting DNA oxidative cleavage method,the inhibitions of CA and its metabolites were studied[24].Mix 100 ng pBR322 plasmid DNA with CA,CFA,and CTA (10,50,100,500,1000 μmol/L),37°Cpreincubatefor10 min,andadd10 mmol/L AAPH to starting reactions.After 37°C for 4 h,the levels of DNA oxidative cleavage were tested by agarose gel electrophoresis,andtheopticaldensitieswerequantifie byQuantity One 4.6.2.
2.11.Cell culture and treatment
Mouse microglial BV2 cells were obtained from the China Center for Type Culture Collection(Wuhan,China).BV2 cells weregrowninRPMI1640mediumwith10%fetalbovineserum,100 μg/mL of streptomycin,and 100 U/mL of penicillin at 37°C under a 5%CO2atmosphere.The medium was replaced every 2 days.BV2 cells were pretreated with different concentrations of CA,CFA,and CTA dissolved in dimethyl sulfoxide for 4 h,and then treated with lipopolysaccharide(1 μg/mL)for 12 h.
2.12.Measurement of cell viability
The cell vitality was determined using MTT assay[25].BV2 cells were seeded in 96-well plates at a density of 1×106cells/well and incubated over night at 37°C with 5%(v/v)CO2.After various treatments,the medium was removed carefully and the cells were incubated with MTT(0.5 mg/mL)for 4 h at 37°C.Formazan crystals formed by live cells were solubilized with 100 μL DMSO and absorbance at 490 nm was measured with a microplate reader(Bio-Rad Laboratories Ltd.,China).Cell viability was expressed as a percentage of the control (untreated cells).The relative cell viability was calculated according to formula(3):whereA1is the absorbance of the experimental group,andA0is the absorbance of the control group.

2.13.Measurement of NO production
The production of NO was determined with Griess reagent[26].50 μL samples or standards were added into 96-well plates,which were incubated with the same volume of the Griess reagent [0.1% (w/v) N-(1-naphathyl)-ethylenediamine and 1%(w/v)sulfanilamide in 5%(v/v)phosphoric acid]at 37°C.Ten minutes later,the absorbance at 540 nm was measured using a microplate reader.The amount of nitrite was calculated by using a sodium nitrite standard curve.
2.14.Measurement of ROS
DCFH-DA fluorescenc dye itself has no fluorescence and can go through the cell membrane freely.However,it can be oxidized into DCF which has fluorescenc by intracellular reactive oxygen species.Therefore,the fluorescenc intensity can reflec the level of ROS[27].

Fig.1.Effects of CA and its metabolites on protein oxidative damage.(a-d)Effects of CA,CTA and CFA on protein oxidative degradation induced by hydroxyl radicals.BSA was incubated with Cu2+/H2O2 in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section;(e-h)Effects of CA,CTA and CFA on protein oxidative degradation induced by alcoxyl radicals.BSA was incubated with AAPH in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section.The protein contents were measured by commasie brilliant blue staining; (i-l) Effects of CA,CTA and CFA on protein carbonylation induced by hydroxyl radicals.BSA was incubated with Cu2+/H2O2 in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section;(m-p)Effects of CA,CTA and CFA on protein carbonylation induced by alcoxyl radicals.BSA was incubated with AAPH in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section.The carbonyl content was determined using a colorimetric method.(q-t)Effects of CA and its metabolites on protein nitration.BSA was incubated with hemin/nitrite/H2O2 in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section; The levels of nitration were measured by western blotting.Each value represents the mean±SD (n ≥3),##represents significan differences compared to the control group(p <0.01);*represents significan differences compared to the induced group(p <0.05);**represents significan differences compared to the induced group (p <0.01); Δ represents significan differences compared to the same concentration of CA (p <0.05); ΔΔ represents significan differences compared to the same concentration of CA(p <0.01).
BV2 microglial cells were washed with PBS 2-3 times after treated,then DCFH-DA dyes diluted by serum free medium were added,after 37°C incubation for 30 min,inverted fluores cence microscope was used to observe the generation of ROS and Multiscan Spectrum to make quantitative determinations ofintracellular ROS.
2.15.Statistical analysis
All the experiments were performed with at least three replications,and all the data were presented as the mean±standard errors (SD).Significan differences were analyzed using the one-way analysis of variance (ANOVA) followed by Tukey’s multiple-range test with the SPSS 18.0 system.
3.Results
3.1.Effects of CA,CTA and CFA on protein oxidative degradation
Excessive generation of ROS will induce protein oxidative damage.And degradation and carbonylation are the main characteristics of protein oxidation [28].Cu2+/H2O2and AAPH reaction systems were adopted to detect the effects of CA and its metabolites on BSA oxidative damage induced by these two different kinds of free radicals.
Fig.1(a-d)showed that after Cu2+/H2O2treatment,protein bands were notably weakened compared to the control group,indicating hydroxyl free radicals produced by Cu2+/H2O2system substantially destroyed the structure of BSA.Pretreatment of CA and CTA increased the content of residual BSA in a concentration dependent manner,which demonstrated that CA and CTA had strong abilities to protect BSA protein from hydroxyl radicals induced degradation.However,the optical density of protein bands in CFA groups decreased.The degradation inhibition rate of 1 mmol/L CA was 60.95%,which was 1.78 times than that of CTA.
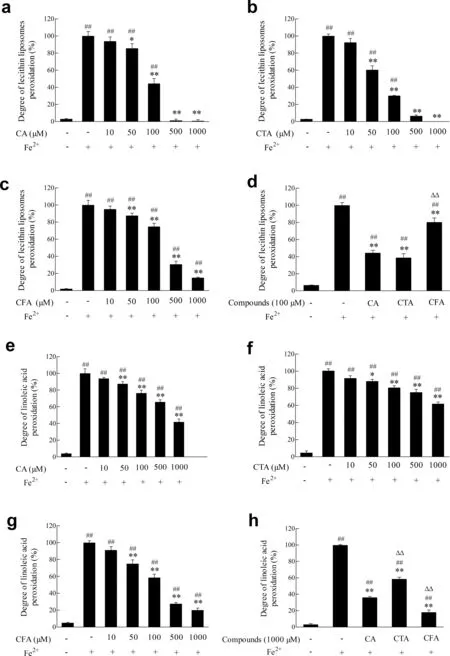
Fig.2.Effects of CA and its metabolites on lipid peroxidation.(a-d)Effects of CA,CTA and CFA on liposomes peroxidation induced by hydroxyl radicals.Lecithin liposomes were incubated with Fe2+ in the absence or presence of CA,CTA and CFA,as described in the Materials and Methods section;(e-h)Effects of CA,CTA and CFA on linoleic acid peroxidation induced by hydroxyl radicals.Linoleic acid was incubated with Fe2+ in the absence or presence of CA,CTA and CFA,as described in the Materials and Methods section.The amount of TBARS formed was determined by colorimetry.Each value represents the mean±SD(n ≥3),##represents significan differences compared to the control group(p <0.01);*represents significan differences compared to the induced group(p <0.05);**represents significan differences compared to the induced group(p <0.01); ΔΔ represents significan differences compared to the same concentration of CA(p <0.01).
AAPH system producing alcoxyl radicals was also employed to investigate the effects of CA and its metabolites on BSA oxidative degradation(Fig.1(e-h)).CA and its metabolites suppressed BSA degradation in concentration-dependent manners.The degradation inhibition rates of CA,CTA and CFA were 79.74%,55.00%and 66.88%at 100 μmol/L,respectively(Supplementary Fig.1).
3.2.Effects of CA,CTA and CFA on protein carbonylation
As illustrated in Fig.1(i-l),Cu2+/H2O2dramatically increased BSA carbonylation.Within the concentration range of 10-1000 μmol/L,CA and its metabolites all exerted remarkable inhibition capacities.Besides,the effects were strengthened along with the increase of the concentrations of CA and CTA.However,CFA promoted carbonylation in 10-1000 μmol/L,which is consistent with the protein degradation at same concentration.The carbonylation suppression rate of 1 mmol/L CA was 37.36%higher than CFA,which had statistically significan difference(p<0.01).
Fig.1(m-p)showedthatthelevelofBSAcarbonylation obviously increased when treated with AAPH,while CA,CTA and CFA all could reverse this trend.The results were consistent with those obtained in BSA degradation system.Carbonylation inhibition rates of CA at 100 μmol/L was 51.46%,dramatically stronger than its metabolites.
3.3.Effects of CA,CTA and CFA on protein nitration
As shown in Fig.1(q-t),treatment with hemin/nitrite/H2O2reaction systemfor60 minsignificantl inducedthe formation of 3-nitrotyrosine in BSA,which suggested obvious BSA protein nitration.In a certain concentration range (10-1000 μmol/L),CAanditsmetaboliteshadsignifican dose-dependentinhibition abilities on nitration induced by hemin/nitrite/H2O2.In addition,500 μmol/L CA and its metabolites pretreatment almost completely restrained the formation of 3-nitrotyrosine.The suppression rates of CA,CTA and CFA at 100 μmol/L were 83.51%,36.09% and 43.99%,respectively (Supplementary Fig.2).Results indicated the stronger inhibiting capacity of CA on hemin/nitrite/H2O2-induced BSA nitration(p<0.01)(Supplementary Fig.2).
3.4.Effects of CA,CTA and CFA on lipid peroxidation
Lipid peroxidation product such as malondialdehyde can react with thiobarbituric acid to generate TBARS,which is an important index in measuring the degree oflipid peroxidation.Lipid had a slow process of auto-oxidation in the absence of Fe2+,and the content of TBARS in induced group increased significantl (p<0.01)compared with control group(Fig.2(a-d)).CA and its metabolites all inhibited the lecithin liposome peroxidation concentration dependently.The ability of 100 μmol/L CA to inhibit liposome peroxidation was 55.60%,which was 2.81 times than that of CFA.However,it had no significan difference compared with CTA(p<0.05).
Moreover,Fig.2(e-h) exhibited a concentration-dependent inhibition in linoleic acid peroxidation when treated with CA and its metabolites,which were consistent with liposome peroxidation system.The inhibition rates of CA at 1 mmol/L was 1.54 and 0.78 times than those of CTA and CFA,respectively.
3.5.Effects of CA,CTA and CFA on DNA oxidative damages
DNA is the main attack target of free radicals.Oxidative stress leads to DNA damages such as DNA base modifica tions,double chain rupture,and base mutation.AAPH thermal decomposition generates alcoxyl radicals,which attack DNA and the degraded product of DNA (reactive carbonyl compounds)willreactwiththiobarbituricacidunderacidiccondition to produce TBARS,which has a significan absorption peak at 532 nm.As demonstrated in Fig.3(a-d),the absorbance of DNA reaction solution at 532 nm significantl (p<0.01) increased after treatment with 40 mmol/L AAPH for 12 h at 37°C.The absorbance of CA and its metabolites treatment group were significantl (p<0.01)lower than AAPH-induced group,indicating that CA and its metabolites all had protective effects on DNA.The degradation suppression rate of 100 μmol/L CA was 72.45%,which was 22.32% and 16.50% statistically significant(p<0.01)higher than those of CTA and CFA,respectively.These results showed that the inhibiting effect of CA on herring sperm DNA induced by alcoxyl radicals was significantl(p<0.01) stronger than its metabolites at same experimental concentrations.
Fig.3(e-h)showed that supercoiled form of DNA unwound and gradually disappeared after the treatment of 10 mmol/L AAPH,while control group was almost all consisted of suprahelical helixes.Pretreatment with 5-100 μmol/L CA and its metabolites increased superhelical structure concentration dependently,implying that CA and its metabolites all remarkably inhibited DNA oxidative cleavage caused by alcoxyl radicals.The content of supercoiled DNA was 82.73% when treated with 100 μmol/L CA,which was significantl (p<0.01)higher than its metabolites CTA and CFA.The result demonstrated that the inhibiting effect of CA on pBR322 plasmid DNA breakage induced by alcoxyl radicals was significantl (p<0.01)stronger than its metabolites.
3.6.Effects of CA,CTA and CFA on BV2 cell viability induced by lipopolysaccharide
Chronic inflammatio plays a pivotal role in pathogenesis of neurodegenerative diseases such as Alzheimer’s disease,Parkinson’s disease and multiple sclerosis.As illustrated in Fig.4,CA and its metabolites themselves had no influenc on cell viability.After treated with lipopolysaccharide (1 μg/mL) for 12 h,viability of BV2 cells significantl decreased to 81.58%compared to control group.However,pretreatment of various concentration of CA and its metabolites improved cell viability compared to lipopolysaccharide treatment group.After pretreatment with 80 μmol/L CA,CTA and CFA,cell viability elevated back to 88.84%,83.64%,and 84.24%,respectively.
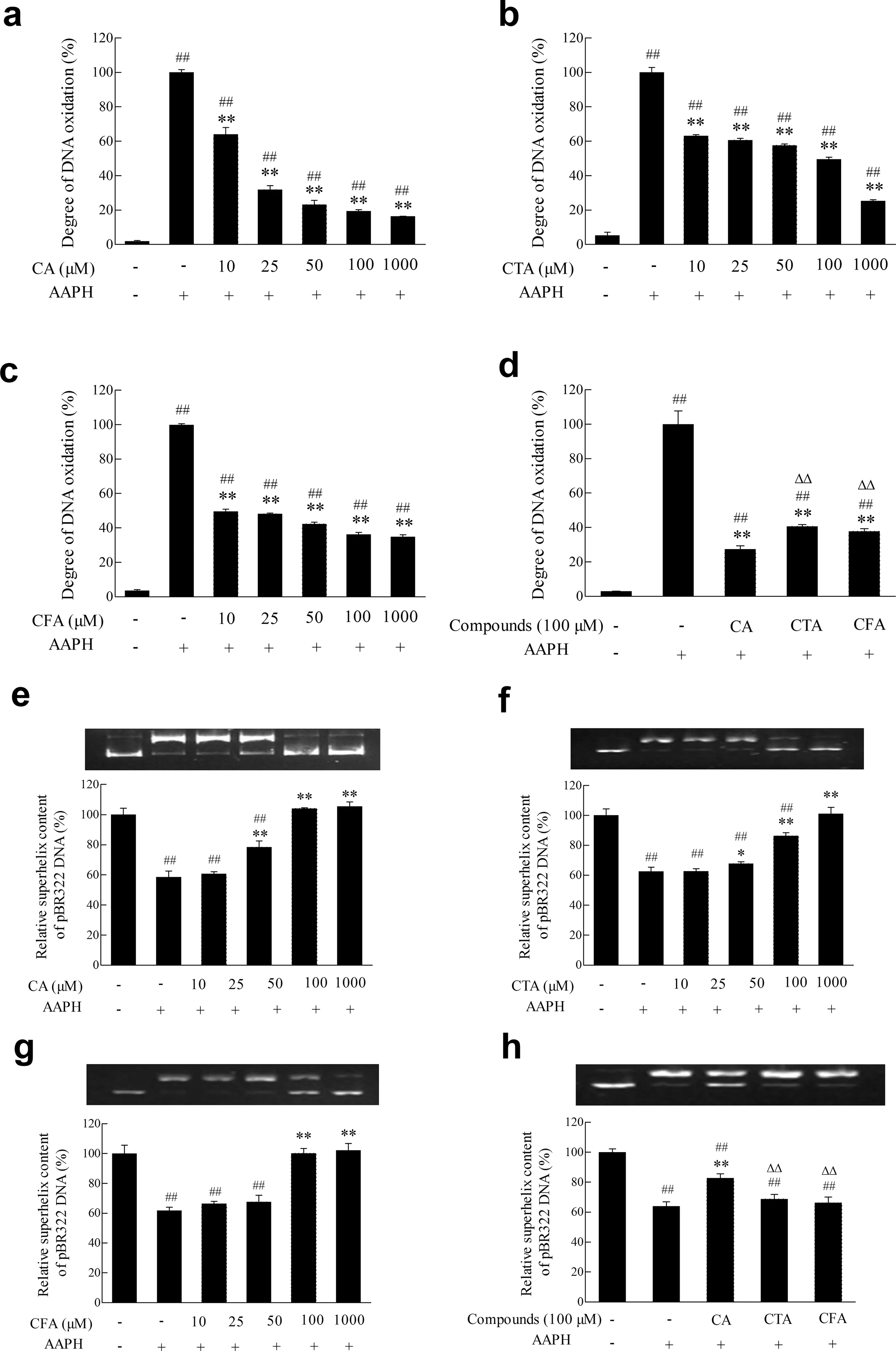
Fig.3.Effects of CA and its metabolites on DNA oxidative damage.(a-d)Effects of CA,CTA and CFA on hsDNA oxidation induced by alcoxyl radicals.hsDNA was incubated with AAPH in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section.The degree of DNA oxidative damage was measured by TBA method; (e-h) Effects of CA,CTA and CFA on pBR322 plasmid DNA oxidative cleavage induced by alcoxyl radicals.pBR322 plasmid DNA was incubated with AAPH in the presence or absence of CA,CTA and CFA,as described in the Materials and Methods section.The levels of DNA oxidative cleavage were tested by agarose gel electrophoresis,and the optical densities were quantifie by Quantity One 4.6.2.Each value represents the mean±SD(n ≥3),##represents significan differences compared to the control group(p <0.01);*represents significan differences compared to the induced group(p <0.05);**represents significan differences compared to the induced group(p <0.01); ΔΔ represents significan differences compared to the same concentration of CA(p <0.01).
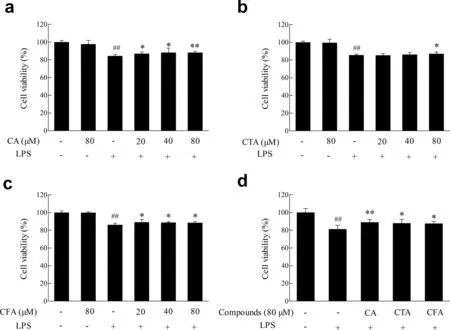
Fig.4.Effect of CA and its metabolites on lipopolysaccharide-induced BV2 cell viability.BV2 cells were treated with lipopolysaccharide(1 μg/mL)12 h with or without of pretreatment of CA,CTA and CFA for 4 h.Cell viability was measured by MTT assay.Each value represents the mean±SD (n ≥8),## represents significan differences compared to the control group(p <0.01);*represents significan differences compared to the induced group(p <0.05);**represents significan differences compared to the induced group(p <0.01).
3.7.Effects of CA,CTA and CFA on the production of NO induced by lipopolysaccharide
NO,an inflammator product,results in biomacromolecule damageandaggravatesinflammator process.Fig.5showedthat CA and its metabolites had no effects on the production of NO in BV2 cells.However,20-80 μmol/L CA and its metabolites remarkably reduced the increased production of NO induced by lipopolysaccharide in concentration-depended manners.The NO inhibition rates of CA,CTA and CFA at 80 μmol/L were 11.14%,10.25%and 9.60%,respectively.
3.8.Effects of CA,CTA and CFA on the production of ROS induced by lipopolysaccharide
ROS is mainly produced by mitochondria and NADPH oxidase located on the cell membrane.Excessive ROS can trigger a variety of signaling pathways such as NFκB and MAPK,leading to cell proliferation,differentiate,or death.As demonstrated in Fig.6,lipopolysaccharide treatment induced elevated ROS levels in BV2 cells.As expected,CA significantl (p<0.01)reduced the ROS levels induced by lipopolysaccharide,which was 7.93 and 2.25 times than the effects of CTA and CFA.
4.Discussion
Various studies have confirme that phytochemicals,especially plant phenols could prevent or alleviate oxidative stress in diseases [29].In this study,antioxidant effects of CA and its metabolites on the protection of biological macromoleculesin vitroand inflammator responses in microglial cells were investigated.Excessive ROS/RNS will attack biological macromolecules such as proteins,lipids and DNA,disrupt cell structure and functions.
Recent studies have found that many diseases such as Alzheimer’s,diabetes,cardiovascular disease are associated with elevated levels of protein carbonylation [30-32].Protein tyrosine nitration has been identifie as an important selective post-translational modification and is vital in the initiation and aggravation in cardiovascular diseases,ischemia/reperfusion injury,inflammation and neurodegenerative disorder.CA,CFA and CTA all significantl inhibited protein degradation and carbonylation induced by hydroxyl radicals and alcoxyl radicals,suppressed the RNS-induced nitration in the experimental concentrations (10-1000 μmol/L).And the protective effects of CA were markedly stronger than its metabolites.It should be noted that CFA promoted oxidation in high concentration range induced by hydroxyl radicals,may due to the reduction of Fe3+to Fe2+,and the reaction between Fe2+and H2O2forming hydroxyl radicals[33].
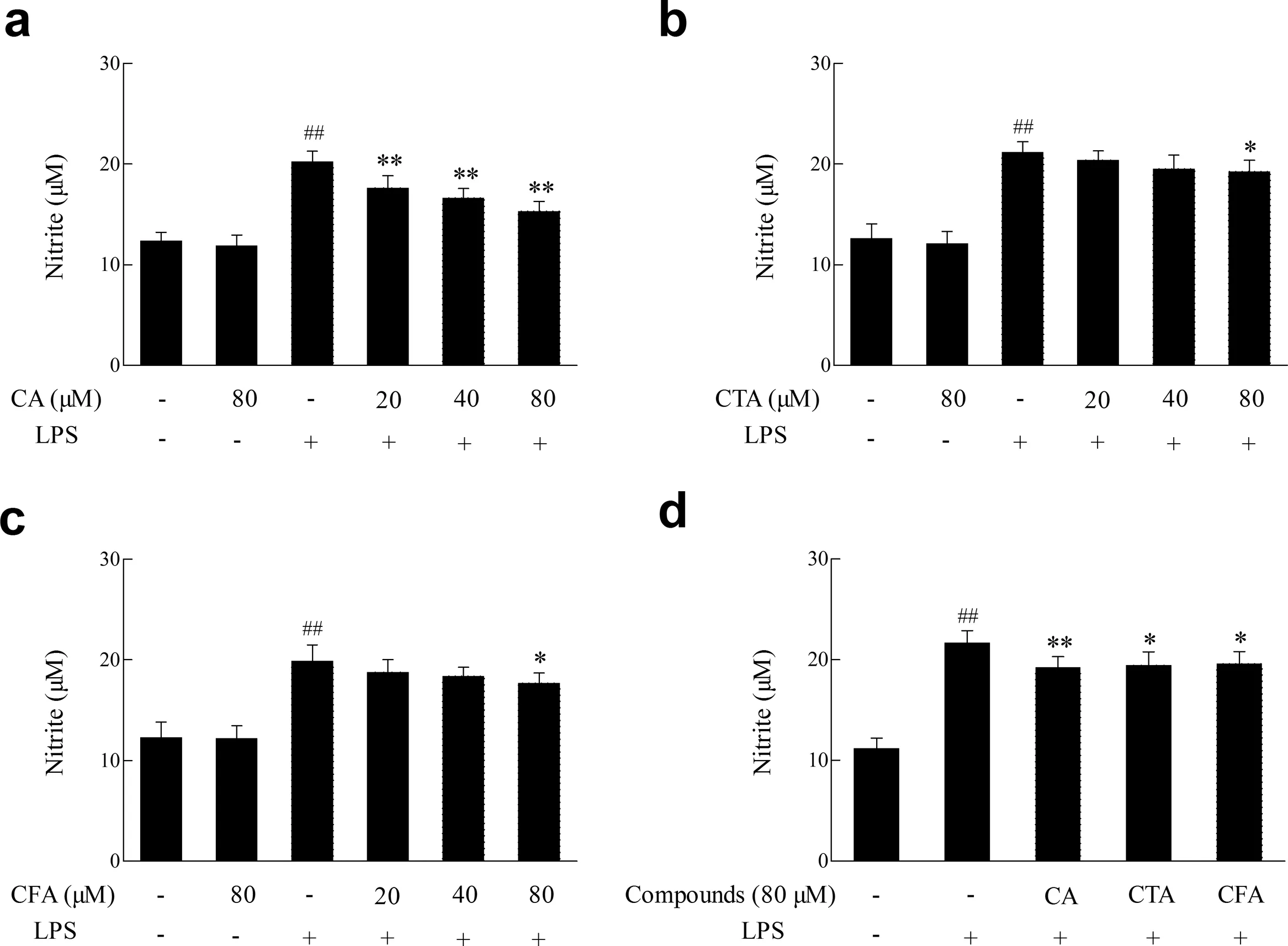
Fig.5.Effect of CA and its metabolites on lipopolysaccharide-induced NO production.BV2 cells were treated with lipopolysaccharide (1 μg/mL) 12 h with or without of pretreatment of CA,CTA and CFA for 4 h.The accumulation of nitrite was measured to assess the NO production in BV2 cells.Each value represents the mean±SD(n ≥8),##represents significan differences compared to the control group(p <0.01);*represents significan differences compared to the induced group(p <0.05); ** represents significan differences compared to the induced group(p <0.01).
Lipid peroxidation readily occurs in mitochondria,cell membrane,endoplasmic reticulum,lysosomes,perioxisomes and other membrane structures that contain rich polyunsaturated fatty acids.In the process oflipid peroxidation,a large number of toxicities including malondialdehyde,acraldehyde and 4-hydroxynonenal are produced[34].Linoleic acid,an essential fatty acid containing two easily oxidized double bonds,is an ideal material for studying lipid peroxidation[35].The polyunsaturated fatty acids of very low density lipoprotein and low density lipoprotein located in the C-2 oflecithin produce peroxidation under the catalysis of Fe2+.So lecithin is usually used as a cell modelin vitro.This study indicated that the inhibition of CA on Fe2+-induced linoleic acid and soybean lecithin liposomes peroxidation were remarkably stronger than its metabolites.It is also worthy of noting that CFA had a better protective effect oflinoleic acid in comparison with CA.
DNA oxidative damage caused by radicals attack is closely related to a variety of diseases such as inflammation tumor,cardiovascular disease and aging.In this work,CA and its metabolites showed a concentration-dependent inhibition on the oxidation of hsDNA and protection for pBR322 plasmid DNA from breakage induced by alcoxyl radicals.Protective effects of CA were statistically significan better than its metabolites.It has been demonstrated that polyphenols in mango wine could protect DNA against UV+H2O2and γ-irradiation induced DNA damage[36].(+)-catechin hydrate,(-)-epigallocatechin gallate hydrate,morin hydrate,quercetin hydrate and resveratrol have potential for protecting both the viability of cells and ability to proliferate from damage caused by UV-A-irradiation [37].Green tea(Camellia sinensis)strongly reversed apoptotic hepatic degeneration and prevented micronecrosis and mutagenic DNA damage induced by arsenic[38].
Chronic inflammatio in the brain is one of the most important pathological features of neural degenerative diseases such as Alzheimer’s,Parkinson’s disease and multiple sclerosis[39].Microglia,the most representative immune cells in the central nervous system,can secrete cytokines such as NO,TNF-α,and IL-1β [40].Lipopolysaccharide is an efficien activator of microglial cells and it can increase the production of ROS and pro-inflammator cytokines,which damage the surrounding nerve cells [41].Lipopolysaccharide activates receptor serine/threonine kinases to trigger the phosphorylation of a series protein kinase through specifireceptor CD14 and TLR4 on the cell membrane,and it also activates p38MAPK and NFκB signalingpathwaystoparticipateintheprocessofiNOSexpression,which promotes the production of NO [42].NO,as an intracellular messenger molecule,takes part in vascular regulation,neurotransmitter,immune responses and other physiological processes [43].However,excess NO will react with superoxide anion (O2·-) to generate peroxynitrite (ONOO-),which decomposes to generate a potent oxidant similar to HO·in reactivity,and it may cross cell membranes through anion channels[44].ONOO-induces protein nitration,lipid peroxidation,and DNA breakage[45].Increased formation of ONOOhas been linked to Alzheimer’s disease,rheumatoid arthritis,atherosclerosis,lung injury,amyotrophic lateral sclerosis,and other diseases [46,47].This study demonstrated that both CA and its metabolites significantl increased BV2 microglial cell viability and inhibited the production of NO and ROS induced by lipopolysaccharide.As noted,the inhibition of CA on the ROS levels induced by lipopolysaccharide was stronger than CTA and CFA,which was consistent with the scavenging of free radicals including DPPH·,·OH,ABTS+·and biological macromolecular protection effectsin vitro.Whereas CA,CTA and CFA had no significan difference in NO levels and cell viability.Since DCFH-DA assay was used to detect overall intracellular redox,cells incubation with compounds with more hydroxyl groups is likely to have a lower DCF fluorescence which may partly contributed to the more powerful inhibitory effects of CA.There are structure-activity relationships of polyphenols to inhibiting lipopolysaccharide-induced inflammatio [48].However,the molecular mechanism remains to be further investigated.
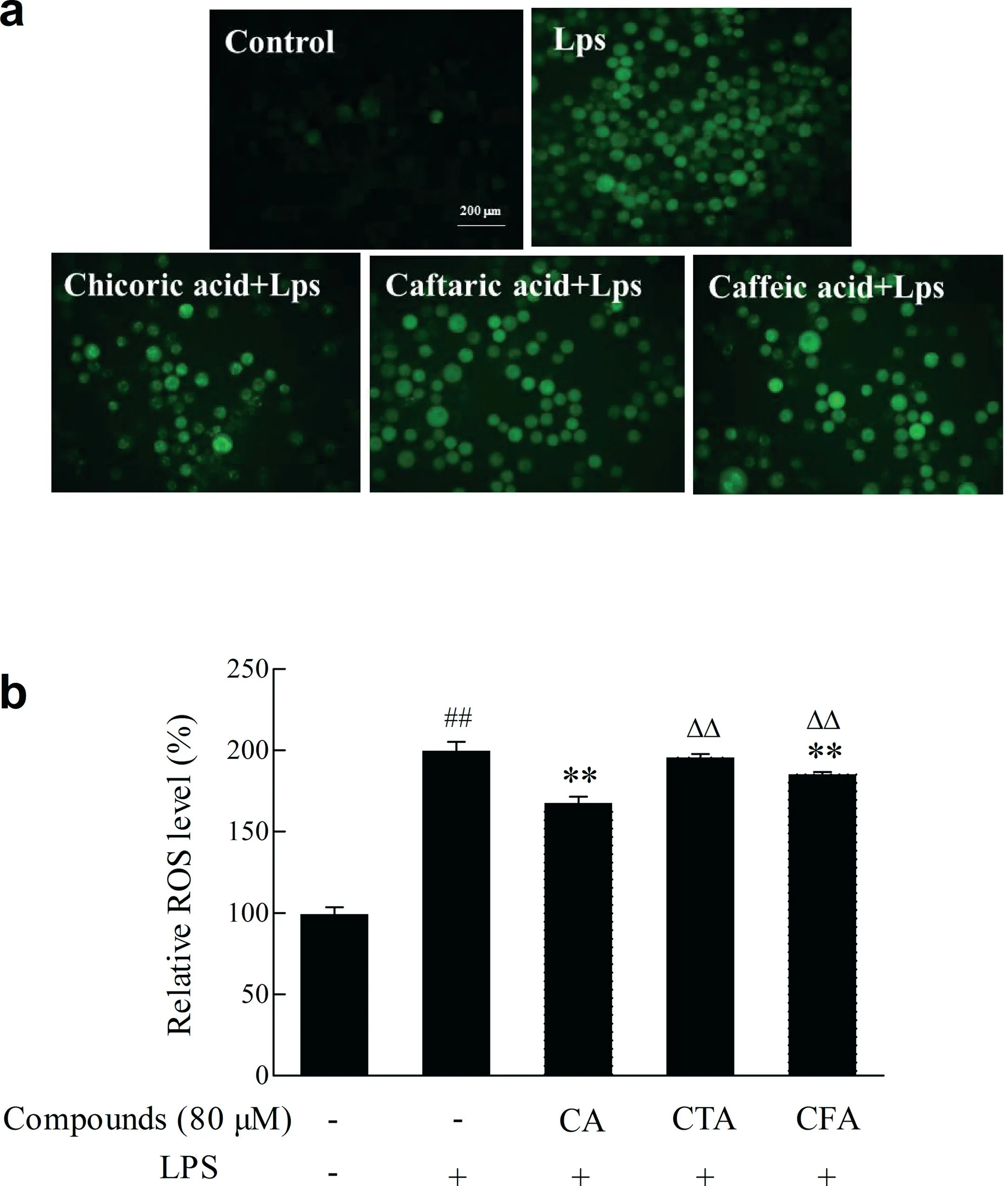
Fig.6.Effect of CA and its metabolites on lipopolysaccharide-induced ROS production.BV2 cells were treated with lipopolysaccharide(1 μg/mL)12 h with or without of pretreatment of CA,CTA and CFA for 4 h.The production of ROS was monitored with DCFH-DA fluorescenc dye.(a) using inverted fluorescenc microscope to observe the generation of ROS; (b) using multiscan spectrum to make quantitative determinations ofintracellular ROS.Each value represents the mean±SD(n ≥8),##represents significan differences compared to the control group(p <0.01);**represents significan differences compared to the induced group(p <0.01); ΔΔ represents significan differences compared to the same concentration of CA(p <0.01).
5.Conclusion
In this study,effects of CA and its metabolites onin vitroprotection of biological macromolecules and inflammator responses in microglial cell were investigated.Results demonstrated that CA and its metabolites all exerted remarkable inhibition capacities to biological macromolecules damages including protein,lipid,and DNA induced by different kinds of free radicals.In addition,CA and its metabolites suppressed the decline of BV2 microglia cell viability and the production of NO and ROS induced by lipopolysaccharide.Furthermore,bioactivities of CA were significantl stronger than those ofits metabolites within a certain concentration range.These find ings will provide scientifibases for the application of CA and its metabolites as nutrition and natural antioxidants.
Conflict ofinterest
The authors declare that there are no conflict ofinterest.
Acknowledgement
This work was supported by the National Key Research and Development Program of China (No.2016YFD0400601) and the Science and Technology Coordination Project of Innovation in Shaanxi province(NO.2014KTCL02-07).
Appendix A.Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.fshw.2017.09.001.

- 食品科学与人类健康(英文)的其它文章
- Effects of Soursop flwers(Annona muricata L.)extract on chemical changes of refine palm olein stored at frying temperature
- About the Beijing Academy of Food Sciences
- Therapeutic molecules for multiple human diseases identifie from pigeon pea(Cajanus cajan L.Millsp.)through GC-MS and molecular docking
- A synthetic biological secondary metabolite,LycogenTM,produced and extracted from Rhodobacter sphaeroides WL-APD911 in an optimizatioal scale-up strategy
- Establishment of probabilistic model for Salmonella Enteritidis growth and inactivation under acid and osmotic pressure
- Terminalia arjuna:A novel natural preservative for improved lipid oxidative stability and storage quality of muscle foods