
雷公藤内酯醇减少放射性肺纤维化中肌成纤维细胞活化与抑制TGF-β1/ERK/Smad3通路相关
2017-05-17 03:51张彦伟张志强吴丽贤张鲁榕
中国药理学通报 2017年5期
张彦伟,张志强,2,3,吴丽贤,2,3,张鲁榕,陈 纯,2,3
(福建医科大学1.药学院药理学系、2.新药研究所、3.福建省天然药物药理学重点实验室,福建 福州 350122;4.福建医科大学附属第一医院, 福建 福州 350004; 5.福建医科大学放射生物学重点实验室,福建 福州 350004)
雷公藤内酯醇减少放射性肺纤维化中肌成纤维细胞活化与抑制TGF-β1/ERK/Smad3通路相关
张彦伟1,张志强1,2,3,吴丽贤1,2,3,张鲁榕4,5,陈 纯1,2,3
(福建医科大学1.药学院药理学系、2.新药研究所、3.福建省天然药物药理学重点实验室,福建 福州 350122;4.福建医科大学附属第一医院, 福建 福州 350004; 5.福建医科大学放射生物学重点实验室,福建 福州 350004)
目的 通过体内外实验,观察雷公藤内酯醇(TPL)减少放射性肺纤维化中肌成纤维细胞(MFBs)活化与TGF-β1/ERK/Smad3通路的相关性。方法 以TGF-β1刺激成纤维细胞建立体外MFBs活化模型,C57BL/6小鼠胸部照射形成体内MFBs活化、放射性肺纤维化模型。MFBs活化状态通过检测小鼠成纤维细胞中α-SMA(RT-PCR、Western blot方法)和Col Ⅰ(RT-PCR、ELISA方法)的表达,通路活性采用Western blot法检测p-ERK、p-Smad3(Ser208)、p-Smad3(Ser423)的水平。ERK siRNA及Smad3 siRNA观察ERK、Smad3在MFBs活化中的地位。结果 TGF-β1激活p-ERK/p-Smad3(Ser208)及p-Smad3(Ser423),诱导成纤维细胞表达α-SMA,活化为MFBs,合成Col I明显增多;使用ERK siRNA及Smad3 siRNA确定了ERK及Smad3均参与α-SMA、Col Ⅰ的表达,其中ERK可能通过磷酸化Smad3连接区(Ser208)起相关作用。小鼠胸部照射可致肺组织中p-ERK、p-Smad3(Ser208)、p-Smad3(Ser423)的水平上调,α-SMA表达增加,显示多量MFBs活化。TPL对体内和体外实验中ERK、Smad3(Ser208)、Smad3(Ser423)的磷酸化活化均有明显抑制作用,可明显下调α-SMA表达及Col Ⅰ合成,减少MFBs的活化。结论 TPL可通过抑制TGF-β1/ERK/Smad3通路,减少肺部照射后MFBs活化,从而抑制放射性肺纤维化进展。
雷公藤内酯醇;放射性肺纤维化;肌成纤维细胞;α-平滑肌肌动蛋白;Ⅰ型胶原蛋白;转化生长因子β1;ERK;Smad3
放射性肺纤维化是胸部肿瘤放射治疗常见而严重的并发症,由于其发生率和严重度与照射剂量及照射范围密切相关,因而成为限制胸部肿瘤(包括发病率高居全国乃至世界前位的肺癌、乳腺癌等)放射剂量的决定性因素,严重干扰肿瘤局部控制率及患者预后。目前,临床对放射性肺纤维化尚无有效措施[1-2]。本研究团队首次发现雷公藤内酯醇(triptolide, TPL)能有效缓解实验性放射性肺纤维化,相应剂量下无明显不良反应,具有良好的开发前景[3]。 有关纤维化的机制研究显示,肌成纤维细胞(myofibroblasts, MFBs)是纤维化病变的核心效应细胞,在增生性疤痕及多种纤维化病变中,MFBs持续活化存在[4-5]。MFBs常由成纤维细胞活化或上皮细胞转化而来,过量的、持续存在的MFBs通过分泌过量胶原蛋白[主要为Ⅰ型胶原蛋白(type Ⅰ collagen, Col Ⅰ)[6]]致细胞外基质过度沉积,通过表达MFBs标志物α-平滑肌肌动蛋白(α-smooth muscle actin, α-SMA)等致组织过度收缩、顺应性下降,最终导致纤维化[4-5,7]。因此,MFBs已成为抗纤维化治疗的重要靶标。
我们的前期研究显示,在放射性肺纤维化中,肺泡巨噬细胞通过分泌大量ROS、以旁分泌途径刺激纤维细胞活化为MFBs,导致纤维化发生、发展;TPL通过抑制肺泡巨噬细胞、下调ROS、阻止MFBs的活化,从而减少基质沉积、缓解纤维化[8]。
但诱导MFBs活化的途径有多种,其中TGF-β1是常见的促MFBs活化、促纤维化的重要因子[9]。在肺组织中,TGF-β1诱导α-SMA(即MFBs活化)主要由Smad3调控[10]。Hayashida等[11]的研究提出,Ras/MEK/ERK参与了对TGF-β1/Smad3通路的调节:TGF-β1与胞膜受体结合后,引起Ras/MEK/ERK的相继激活, 活化的ERK继而引起Smad3连接区丝/苏氨酸的磷酸化(Ser 208/212);另一方面,TGF-β1诱导Smad3 C端磷酸化(Ser 423/425,为Smad3入核的基础),此多部位磷酸化的Smad3(Ser208/212, Ser423/425)与Smad4结合后入核,调节相关基因转录的能力增强[12],促进纤维细胞增生及胶原蛋白沉积[13]。但ERK是否通过磷酸化Smad3进而参与调节α-SMA的表达,目前尚无相关研究。 在放射性肺纤维化发生过程中持续高表达TGF-β1,且TGF-β1水平可用于预测放疗后发生肺纤维化的危险性[14],可见TGF-β1在放射性肺纤维化的发生中起重要作用,是促MFBs活化的重要因素之一。TPL为多靶点作用药物,且前期结果显示TPL具有强效抗放射性肺纤维化作用,因此TPL对MFBs的抑制作用可能亦为多途径。本实验观察TPL对TGF-β1下游信号分子ERK、Smad3的影响,证实了TPL可通过抑制ERK、Smad3的磷酸化激活,减少MFBs活化。
1 材料与方法
1.1 药物 雷公藤内酯醇(TPL)纯品购自上海柘智生物科技有限公司,纯度>99%。TPL的配制:1 mg TPL加入1 mL DMSO充分溶解后,以注射用生理盐水定容至10 mL,即配制成100 mg·L-1的浓缩液,分装。用时以注射用生理盐水稀释至1 mg·L-1,微孔滤膜(0.22 μm)过滤除菌。
1.2 试剂 UltraCULTURETM无血清培养基(LONZA, USA);DMEM培养液、胰蛋白酶(Gibco,USA);胎牛血清(FBS,四季青生物工程技术研究所);TGF-β1(R&D Systems,USA);Collagen Ⅰ ELISA 试剂盒(DuoSet ELISA kits,R&D Systems);BIOZOL Total RNA Extraction Reagent(BioFlux,Germany);BioRT cDNA第一链合成试剂盒、BioEasy SYBR Green Ⅰ RT-PCR Kit(杭州博日科技); 一抗anti-α-SMA、anti-phospho-ERK1/2、anti-ERK1/2(Abcam,USA);anti-phospho-Smad3(Ser208)、anti-phospho-Smad3(Ser423)、anti-Smad3(Cell Signaling Technology, USA);SignalSilence®p44/42 MAPK(ERK1/2) siRNA(Cell Signaling Technology);si-m-Smad3(锐博生物科技有限公司);X-tremeGene siRNA transfection reagent (Roche Diagnostics Corp., USA) ;青霉素-链霉素溶液(100×)、BCA蛋白浓度测定试剂盒、辣根过氧化物酶标记二抗、ECL试剂(碧云天生物技术有限公司)。
1.3 方法
1.3.1 细胞培养 NIH3T3(小鼠成纤维细胞株)细胞购自中科院上海细胞所。以含5% FBS的DMEM培养液于37℃、5% CO2常规培养、传代、冻存。收集NIH3T3细胞,以含0.01% FBS的DMEM液饥饿培养12 h后进入实验,采用UltraCULTURETM无血清培养液,以生理盐水、TPL预处理6 h后,加入TGF-β1(5 μg·L-1)刺激NIH3T3细胞1、2 d,分别收集细胞及培养液。
1.3.2 RT-PCR检测 引物由上海生工生物工程有限公司设计合成。收集细胞,以预冷的PBS洗涤3次,吸干水分,以Biozol总RNA提取试剂提取细胞总RNA。NanDrop 2000超微量分光光度计检测所有RNA样品的OD260/OD280吸光度比值(R值)均在1.8~2.2。根据BioRT cDNA第一链合成试剂盒说明书于冰上配制RNA逆转录反应体系,加入RNA样品,使用T100TMThermal Cycler基因扩增仪进行逆转录反应,合成cDNA。根据BioEasy SYBR Green Ⅰ RT-PCR Kit说明书,于冰上配制反应体系(25 μL),在实时荧光定量PCR仪进行PCR反应。反应条件:94℃ 预变性2 min;94℃ 变性10 s,57℃退火20 s,40个循环。以GADPH作为内参,于72 ℃采集荧光信号,采用2-△△Ct法计算对照组和照射组中的表达水平差异。实验重复3次。引物序列见Tab 1。
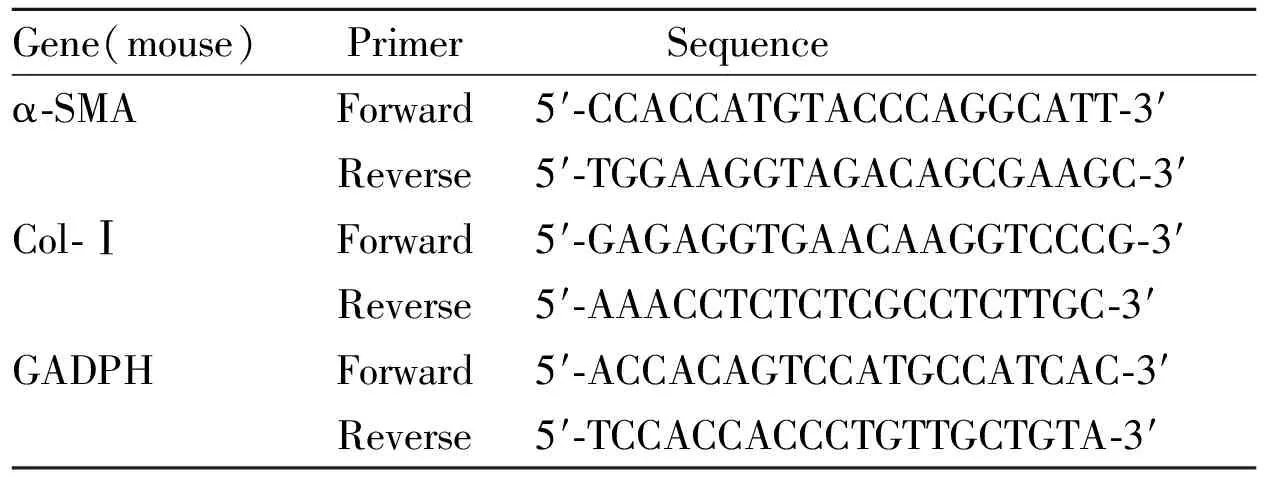
Tab 1 Primer sequence
1.3.3 ELISA法检测Col Ⅰ含量 采用Collagen Ⅰ ELISA试剂盒,按说明书进行实验。以包被缓冲液稀释Collagen Ⅰ抗体,每孔加0.1 mL,4℃包被过夜。洗涤缓冲液洗3次,每次3 min;加5%小牛血清白蛋白置37℃封闭40 min,洗涤1次3 min;将收集的细胞培养液0.1 mL加入孔中,37℃孵育1 h后,洗涤3次;于各反应孔中加入新鲜稀释的酶标抗体0.1 mL,37℃孵育0.5~1 h,洗涤3次;其后加新鲜配制的TMB底物溶液0.1 mL,37℃反应10~30 min,加入2 mol·L-1硫酸0.05 mL终止反应。酶标检测仪(EnVision 1900,PerkinElmer,USA)检测OD450 nm值。根据标准曲线即可得各孔样本浓度。
1.3.4 siRNA抑制ERK、Smad3 NIH3T3细胞在含5% FBS的DMEM培养液(无抗生素)中生长至60%汇合,然后分别转染nontargeting对照siRNA、si-m-Smad3 (duplex1,20 nmol·L-1)或 ERK siRNA(50 nmol·L-1),采用X-tremeGene siRNA transfection reagent转染试剂。通过检测对照siRNA(Cy3标记)的荧光值(Cellomics Arrayscan VT1, Thermo Scientific, USA),确定转染效率为80%~85%。转染细胞的培养及处理同普通NIH3T3。
1.3.5 Western blot检测蛋白含量 收集的细胞以预冷PBS洗涤3次,加入200~300 μL的RIPA裂解液,于冰上裂解30 min,期间反复震荡3~4次;4℃、10 000 r·min-1离心10 min,吸取上清(即总蛋白),BCA法测定蛋白含量;上清中加4×SDS上样缓冲液,沸水浴5 min,-20℃保存。制备聚丙烯酰胺凝胶(分离胶:10%),每孔蛋白上样量约40 μg,电泳分离样本蛋白质。以300 mA湿法电转至PVDF膜,将膜置于5%小牛血清白蛋白中封闭1 h;一抗[anti-α-SMA、anti-phospho-ERK1/2、anti-ERK1/2、anti-phospho-Smad3(Ser208)、anti-phospho-Smad3(Ser423)、anti-Smad3]用封闭液稀释后覆盖PVDF膜上于4℃孵育过夜,TBST洗3次,每次5 min;将1 ∶5 000稀释的二抗加于PVDF膜上,室温下孵育2 h,TBST清洗3次,每次5 min;其后加入ECL试剂显色,以Image Station 4000MM成像仪(DL Naturegene Life Sciences,USA)成像。蛋白条带灰度值采用ImageJ软件分析。
-20℃冻存的小鼠肺组织样本剪成碎块,置于预冷RIPA裂解液中(每克组织加3mL RIPA液),于冰上匀浆并裂解30 min,其后操作同细胞样本。
1.3.6 小鼠肺照射模型 C57BL/6小鼠[♀,8周龄,上海斯莱克实验动物有限公司,许可证号SCXK(沪)2012-0002],氯胺酮麻醉(80 mg·kg-1,ip),以特制夹具保证小鼠肺处在辐射场中,15 Gy全肺照射。照射小鼠随机分组,每组8只,分为对照组(生理盐水)和TPL组(0.25 mg·kg-1, iv,隔日1次),给药1个月(给药剂量及疗程由前期实验确定[8])。设正常小鼠平行对照。照射后4~5个月出现放射性肺纤维化[3],本实验于照射后35~37 d终止实验,颈椎脱臼处死小鼠,取肺组织样本进行相关测定。其中约1/4肺组织低温匀浆(每克组织加3 mL PBS液),以ELISA法检测TGF-β1 、Col Ⅰ含量;余下部分用于Western blot检测(见“1.3.5”项)。所有动物相关实验均通过福建医科大学动物实验福利伦理委员会批准。
2 结果
2.1 TPL减少TGF-β1诱导的MFBs活化 经TGF-β1(5 μg·L-1)诱导后,NIH3T3细胞中α-SMA及Col Ⅰ的转录及表达均明显增加(Pro-Col Ⅰ为Col Ⅰ未交联前的可溶态),即TGF-β1促进成纤维细胞活化为MFBs,建立了体外促MFBs活化的模型。TPL 2.5、5 μg·L-1可明显抑制α-SMA及Col Ⅰ的转录,下调α-SMA水平,减少Pro-Col Ⅰ的分泌,从而抑制TGF-β1诱导的MFBs活化(Fig 1)。
2.2 TPL抑制MFBs形成与抑制ERK/Smad3(Ser208)和Smad3(Ser423)的磷酸化活化有关 Western blot结果显示(Fig 2A、2B),TGF-β1诱导可上调成纤维细胞中p-ERK、p-Smad3(Ser208)、p-Smad3(Ser423)水平,显示ERK/Smad3信号通路的激活。
TGF-β1/Smad3通路是促α-SMA表达、Col Ⅰ合成(即MFBs活化)的重要途径。本实验利用siRNA观察ERK对TGF-β1/Smad3的调节作用,以及ERK、Smad3在MFBs活化中的地位。Fig 3B显示,ERK siRNA可抑制Smad3(Ser208)的磷酸化,而对p-Smad3(Ser423)水平无明显影响,表明经TGF-β1激活的ERK可致Smad3连接区磷酸化。ERK siRNA及Smad3 siRNA均可明显抑制TGF-β1刺激的α-SMA表达和Col Ⅰ合成,减少MFBs活化。siRNA结果表明,ERK可通过磷酸化Smad3连接区Ser208,参与调节Smad3的功能,从而部分影响MFBs活化;Smad3 siRNA则直接抑制Smad3的活性,明显减少MFBs活化(Fig 3C、3D)。
经TPL(2.5、5 μg·L-1)预处理后,TGF-β1诱导的ERK、Smad3(Ser208)、Smad3(Ser423)磷酸化均明显受抑制(Fig 2A、2B)。综上,抑制TGF-β1下游ERK的活化,从而下调p-Smad3(Ser208),以及减少p-Smad3(Ser423)磷酸化,是TPL减少TGF-β1诱导的MFBs活化的重要途径。
2.3 TPL对照射后小鼠肺组织中TGF-β1/ERK/Smad3通路的影响 放射性肺纤维化的形成需4~5个月。在肺部照射1个月后,TGF-β1含量较正常对照组明显升高(数据未显示),α-SMA表达增多(Western blot结果见Fig 4A,流式及免疫组化结果见已发表数据[8]),显示存在多量MFBs活化,此时Col Ⅰ的合成尚未明显增多(Fig 4B),无明显基质沉积[8],表明放射性肺纤维化尚处在发生的早期,活化形成的MFBs分泌的Col Ⅰ尚未累积到一定量。检测TGF-β1/ERK/Smad3通路状况,显示p-ERK、p-Smad3(Ser208)、p-Smad3(Ser423)水平均明显上调(Fig 4C、4D)。
TPL 0.25 mg·kg-1静脉给药1个月,可明显减少照射后肺组织TGF-β1含量,抑制ERK、Smad3(Ser208)、Smad3(Ser423)的磷酸化活化,下调α-SMA表达,即减少MFBs的活化(Fig 4)。动物实验结果证实了TPL可通过抑制TGF-β1/ERK/Smad3途径,减少MFBs数量,从而抑制纤维化的进展。
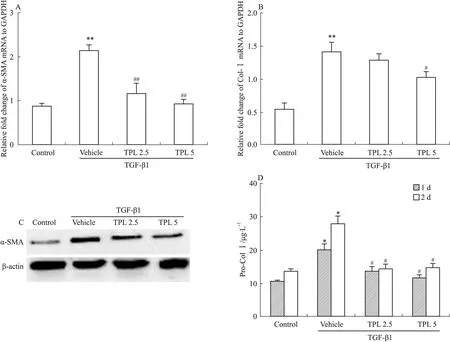
Fig 1 TPL suppressed activation of MFBs induced by TGF-β1
A:α-SMA mRNA in NIH3T3 cells assayed by RT-PCR;B:Col Ⅰ mRNA in NIH3T3 cells assayed by RT-PCR;C:The protein level of α-SMA detected by Western blot;D:The quantity of Col Ⅰ in culture medium measured by ELISA. TPL 2.5 and TPL 5 reflex TPL 2.5 μg·L-1and 5 μg·L-1.*P<0.05,**P<0.01vscontrol;#P<0.05,##P<0.01vsvehicle
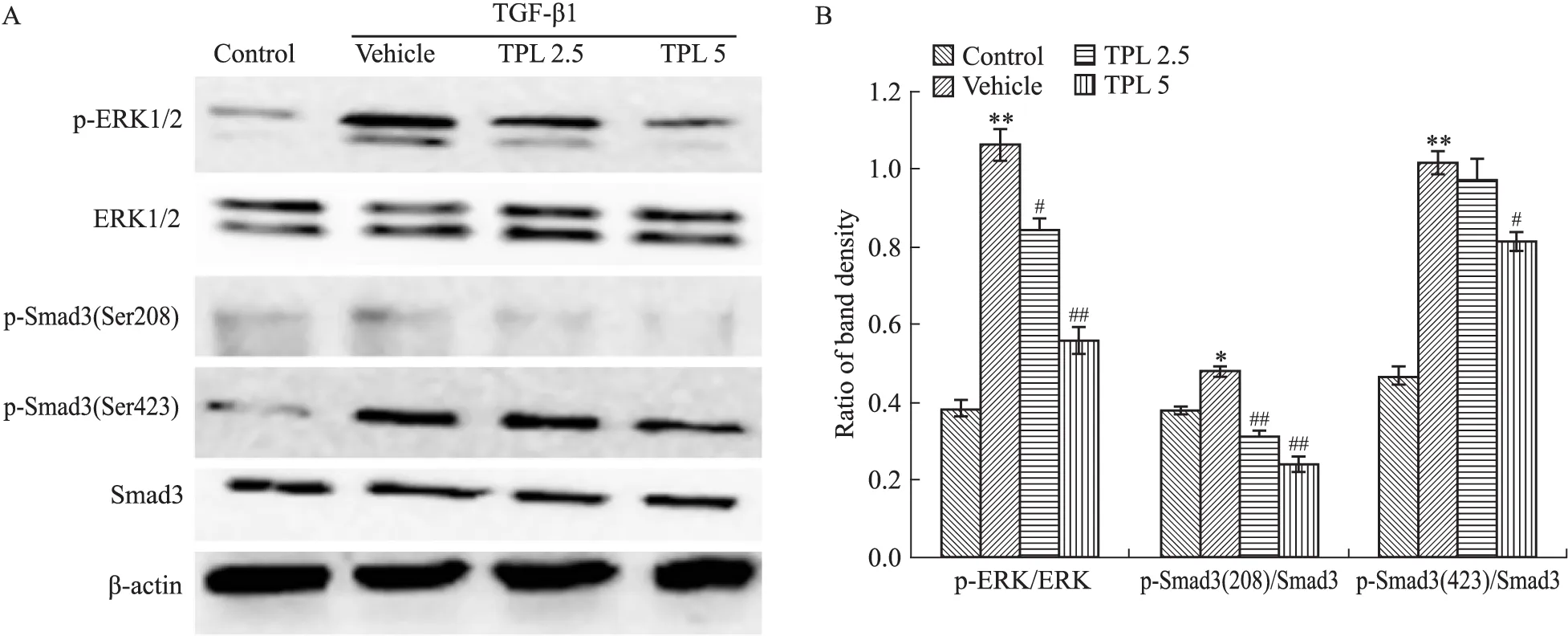
Fig 2 TPL regulated TGF-β1/ERK/Smad3 pathway
A:The phosphorylations of ERK and Smad3 detected by Western blot;B:The protein band density recognized by ImageJ software, and the results presented as ratio.TPL 2.5 and TPL 5 reflex TPL 2.5 μg·L-1and 5 μg·L-1.*P<0.05,**P<0.01vscontrol;#P<0.05,##P<0.01vsvehicle
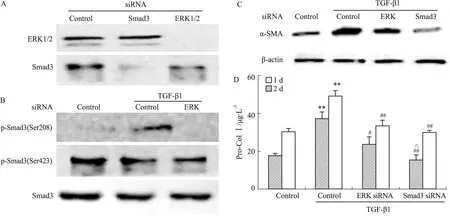
Fig 3 ERK siRNA and Smad3 siRNA inhibited transformation of MFBs from fibroblasts
A:ERK siRNA and Smad3 siRNA effectively knockdowned the expression of ERK and Smad3 respectively;B:Effect of ERK siRNA on the protein level of p-Smad3(Ser208) and p-Smad3(Ser423);C:ERK siRNA and Smad3 siRNA downregulated the level of α-SMA;D:ERK siRNA and Smad3 siRNA reduced the synthesis of Col Ⅰ.**P<0.01vscontrol without TGF-β1;#P<0.05,##P<0.01vscontrol with TGF-β1;△P<0.05vsERK siRNA with TGF-β1
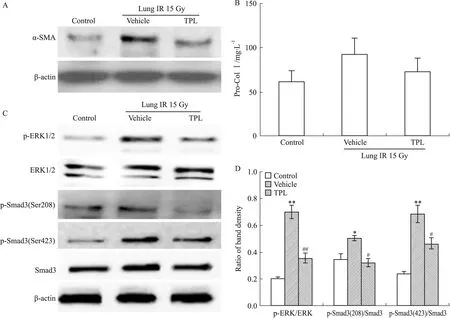
Fig 4 TPL suppressed MFBs activation and TGF-β1/ERK/Smad3 pathway in lung tissue after radiation for 1 month
A:The level of α-SMA in lung tissue measured by Western blot;B:The quantity of Col I in lung tissue assayed by ELISA;C:The phosphorylation of ERK and Smad3 detected by Western blot;D:The protein band density analyzed by ImageJ software, and the results presented as ratio.*P<0.05,**P<0.01vscontrol;#P<0.05,##P<0.01vsvehicle
3 讨论
本实验采用体外、体内实验证实TPL可抑制放射性肺纤维化中TGF-β1诱导的MFBs活化。多种细胞因子可促进MFBs的活化,如TGF-β1、ROS、CTGF、PDGF等,而这些因子主要来源于巨噬细胞、淋巴细胞及MFBs本身[6,15]。前期研究发现,照射后肺组织中肺泡巨噬细胞以旁分泌形式刺激邻近的成纤维细胞活化为MFBs,TPL可下调肺泡巨噬细胞分泌的ROS、减弱其旁分泌作用,减少MFBs的活化[8]。本实验进一步观察TPL对常见促MFBs活化的TGF-β1/Smad3途径的影响。结果表明,TPL可抑制成纤维细胞及肺组织中的Smad3磷酸化激活,减少MFBs经TGF-β1途径活化,并下调Col I的分泌量,减少基质沉积,抑制纤维化的进展。综合我们的结果显示,TPL的抗放射性肺纤维化作用为多靶点效应,对MFBs的活化具有多水平阻断作用:既具有细胞水平的调节作用(巨噬细胞),也表现分子水平的阻断作用。同时,在分子水平具有多靶点作用,可抑制促纤维化因子ROS以及TGF-β1的作用,减少MFBs的活化。
TPL减少照射后早期MFBs的形成,可抑制纤维化的缓慢发展。放射性肺纤维化是慢性发生过程,小鼠放射性肺纤维化的形成需4~5个月,而TPL(0.25 mg·kg-1, iv隔日1次)给药1个月后停药,可以明显缓解其后肺组织的结构变化[3]。结合前期实验[8]及本实验相关机制研究的结果显示,TPL通过其多水平、多靶点的强效抑制作用,明显减少照射后MFBs的形成,从而减弱基质沉积、结构紊乱、顺应性下降等病理改变,达到缓解肺纤维化的效果。
本实验提出ERK可能通过调节Smad3连接区磷酸化参与调节α-SMA的表达。TGF-β1/Smad2/3通路介导了复杂多样的甚至对立的功能调节,如既有促进干细胞多能性,又有刺激干细胞分化的作用等[16]。目前已了解TGF-β1/Smad2/3通路的效应受到多因子、多水平的调节,使得在特定的细胞类型、细胞外环境下引起特定的基因表达、蛋白合成,参与相关的生理或病理过程[16]。在促进纤维细胞增生、Col Ⅰ表达、合成的过程中,TGF-β1/Smad2/3通路主要受到ERK的调节[13,17-18]。由于纤维细胞的增生、合成Col Ⅰ能力增强是纤维细胞活化为MFBs的重要表现,故本实验观察ERK对MFBs活化标志物α-SMA表达的影响。使用siRNA阻断ERK活性,可下调α-SMA的表达及Col Ⅰ的合成,显示ERK确实参与了α-SMA的表达和MFBs的活化。同时,ERK siRNA仅下调p-Smad3(Ser208)水平,而对Smad3(Ser423)的磷酸化无明显影响,表明ERK对α-SMA表达水平的影响可能与ERK磷酸化Smad3连接区(Ser208),从而增强C端磷酸化的Smad3(Ser423)的促基因转录作用有关。进一步确定ERK对TGF-β1/Smad3途径的调节,还需在Smad3敲除的成纤维细胞中过表达ERK,观察成纤维细胞的活化水平。
本实验首次报道TPL的抗放射性肺纤维化作用与抑制TGF-β1/ERK/Smad3途径,减少MFBs活化部分相关。并提出ERK可能通过磷酸化Smad3连接区参与调节Smad3功能,影响α-SMA、Col I的表达,进而影响MFBs的活化。我们的研究显示,TPL在放射性肺纤维化应用方面极具潜力。
(致谢:本实验主要在福建医科大学福建省天然药物药理学重点实验室及福建医科大学附属第一医院放射生物学重点实验室完成,在此对实验室的各位老师及同学的帮助致以衷心的感谢!)
[1] Dicarlo A L,Jackson I L,Shah J R, et al. Development and licensure of medical countermeasures to treat lung damage resulting from a radiological or nuclear incident[J].RadiatRes, 2012,177(5):717-21.
[2] Wynn T A, Ramalingam T R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease[J].NatMed, 2012, 18(7): 1028-40.
[3] Yang S M, Zhang M,Chen C, et al. Triptolide mitigates radiation-induced pulmonary fibrosis[J].RadiatRes, 2015, 184(5):509-17.
[4] Abraham D J, Eckes B, Rajkumar V, Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma[J].CurrRheumatolRep, 2007, 9(2):136-43.
[5] Yarnold J, Brotons M C. Pathogenetic mechanisms in radiation fibrosis[J].RadiotherOncol, 2010, 97(1):149-61.
[6] Friedman S L. Mechanisms of hepatic fibrogenesis[J].Gastroenterology, 2008,134(6):1655-69.
[7] Barnes J L, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases[J].KidneyInt, 2011, 79(9):944-56.
[8] Chen C, Yang S M, Zhang M, et al.Triptolide mitigates radiation-induced pulmonary fibrosis via inhibition of axis of alveolar macrophages-NOXes-ROS-myofibroblasts[J].CancerBiolTher, 2016,17(4):381-9.
[9] Klingberg F, Chow M L, Koehler A, et al. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation[J].JCellBiol, 2014, 207(2):283-97.
[10]Reisdorf P, Lawrence D A, Sivan V, et al. Alteration of transforming growth factor-beta1 response involves down-regulation of Smad3 signaling in myofibroblasts from skin fibrosis[J].AmJPathol, 2001, 159(1): 263-72.
[11]Hayashida T,Decaestecker M, Schnaper H W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells[J].FASEBJ, 2003, 17(11):1576-8.
[12]Blanchette F, Rivard N, Rudd P, et al. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation[J].JBiolChem, 2001, 276(36): 33986-94.
[13]Li F, Fan C, Cheng T, et al. Efficient inhibition of fibroblast proliferation and collagen expression by ERK2 siRNAs[J].BiochemBiophyResCommun, 2009,382(2):259-63.
[14]Stone H B, Coleman C N, Anscher M S,et al. Effects of radiation on normal tissue: consequences and mechanisms[J].LancetOncol, 2003, 4(9): 529-36.
[15]Wynn T A. Cellular and molecular mechanisms of fibrosis[J].JPathol, 2008, 214(2):199-210.
[16]Massague J. TGFbeta signalling in context[J].NatRevMolCellBiol, 2012,13(10): 616-30.
[17]Hayashida T, Wu M H, Pierce A, et al. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397[J].JCellSci, 2007, 120(Pt 23): 4230-40.
[18]Ponticos M, Holmes A M, Shi-wen X, et al. Pivotal role of connective tissue growth factor in lung fibrosis:MAPK-dependent transcriptional activation of type Ⅰ collagen[J].ArthritisRheum, 2009, 60(7):2142-55.
TPL’s suppression on activation of myofibroblasts in radiation induced lung fibrosis related to its inhibition on TGF-β1/ERK/Smad3 pathway
ZHANG Yan-wei1, ZHANG Zhi-qiang1,2,3, WU Li-xian1,2,3, ZHANG Lu-rong4,5, CHEN Chun1,2,3
(1.DeptofPharmacology,2.InstituteofMateriaMedica,3.FujianKeyLaboratoryofNaturalMedicinePharmacology,FujianMedicalUniversity,Fuzhou350122,China;4.theFirstAffiliatedHospitalofFujianMedicalUniversity,Fuzhou350004,China;5.KeyLaboratoryofRadiationBiology,FujianMedicalUniversity,Fuzhou350004,China)
Aim To observe the correlation between the TPL’s suppression on myofibrolbasts(MFBs) activation and TGF-β1/ERK/Smad3 pathway by performinginvivoandinvitroexperiments.MethodsInvitromodel of MFBs activation was set up by stimulating fibroblasts with TGF-β1, andinvivomodel of MFBs activation in radiated lung tissue was built by thoracic radiation on C57BL/6 mice. MFBs activation was analyzed by detecting the expression of α-SMA(using RT-PCR and Western blot) and Col Ⅰ(using RT-PCR and ELISA methods). The levels of p-ERK, p-Smad3(Ser208) and p-Smad3(Ser423) were measured by Western blot. ERK siRNA and Smad3 siRNA were used to observe the status of ERK and Smad3 in MFBs activation.Results TGF-β1 activated p-ERK/p-Smad3(Ser208) and p-Smad3(Ser423), increased the expression of α-SMA and synthesis of Col Ⅰ, which indicated MFBs activation. siRNA knockdown assay showed that both ERK and Smad3 were involved in regulating the levels of α-SMA and Col Ⅰ, and ERK influenced MFBs transformation possibly through its phosphorylation of Smad3(Ser208). TPL treatment inhibited the phosphorylation activation of ERK, Smad3(Ser208), Smad3(Ser423)invitroandinvivo, therefore significantly reduced the level of α-SMA and Col Ⅰ, and the number of activated MFBs was decreased.Conclusion TPL mitigates radiation-induced pulmonary fibrosis by inhibiting the activation of MFBs, which is partly through suppressing TGF-β1/ERK/Smad3 pathway.
triptolide;radiation induced pulmonary fibrosis; myofibroblast; α-smooth muscle actin; type Ⅰ collagen;transforming growth factor β1(TGF-β1); ERK; Smad3
2017-02-14,
2017-03-13
国家自然科学基金资助项目(No 81473264);福建省卫生厅中医药科研项目(No wzze201308);福建省自然科学基金面上项目(No 2014J01336)
张彦伟(1994-),女,硕士生,研究方向:天然药物药理学,E-mail: 2453181338@qq.com; 陈 纯(1971-),女,博士,副教授,硕士生导师,研究方向:天然药物药理学、放射保护药理学,通讯作者,Tel:0591-22862016, E-mail:chenchun-0428@163.com
时间:2017-4-24 11:20
http://kns.cnki.net/kcms/detail/34.1086.R.20170424.1120.018.html
10.3969/j.issn.1001-1978.2017.05.009
A
1001-1978(2017)05-0630-07
R329.25;R563.102.2;R563.130.22;R818;R977.6
猜你喜欢
中老年保健(2022年2期)2022-11-25
皮肤性病诊疗学杂志(2022年3期)2022-08-01
昆明医科大学学报(2022年4期)2022-05-23
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年4期)2021-07-23
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
分析化学(2017年12期)2017-12-25
