
NGO/NC复合含能材料的制备及热分解性能
2017-05-08 01:04李兆乾段晓惠罗庆平裴重华
含能材料 2017年3期
袁 申, 李兆乾, 段晓惠, 罗庆平, 刘 勋, 裴重华
(西南科技大学四川省非金属复合与功能材料重点实验室——省部共建国家重点实验室培育基地, 四川 绵阳 621010)
1 引 言
固体推进剂的燃烧性能是核心技术之一,燃烧性能对固体发动机所产生的推力有着关键性影响[1-2]。当前,研究人员一般采用添加燃烧催化剂来改善固体推进剂的燃烧性能[3-4]。传统的燃烧催化剂已在固体推进剂中得到广泛的应用,但由于其反应活性低,使得催化性能不理想,远不能满足现代军工事业的发展要求[5]。因此,对新型燃烧催化剂的研究显得尤为重要。碳材料作为燃烧催化剂在固体推进剂中的应用已有多年历史[6-7],随着新型碳材料的出现及其特殊结构所展现出优良的性能,使得新型碳材料对固体推进剂催化性能的研究成为了热点。氧化石墨烯(GO)作为新型碳材料燃烧催化剂,在燃烧时,其亲核含氧基团将产生很多活性位点,从而表现出了较好的催化性能[8]。Zhang 等[9]利用溶剂-非溶剂法制备了GO/硝化纤维素(NC)复合材料,研究了GO对NC热分解过程的影响; Li 等[10]将GO与奥克托今(HMX)进行复合,研究了GO对HMX燃烧性能的影响。近年来,燃烧催化剂已向含能燃烧催化剂发展。含能燃烧催化剂的反应活性高、所占比重小,而且其中有大量的含能基团如硝基(—NO2)存在,使其具有高的密度和生成焓,因此含能燃烧催化剂具有较高的能量水平[11-12]。利用—NO2部分取代GO上大量的羟基、羧基和环氧基团等亲核含氧基团,从而得到含能燃烧催化剂硝化石墨烯(NGO)。NGO中不仅有—NO2这种含能基团的存在,而且部分取代使NGO仍具有亲核含氧基团,因此NGO在调节固体推进剂燃烧性能的同时,还能改善其能量水平。Zhang 等[13]用NGO与高氯酸铵(AP)进行复合,研究了NGO对AP热分解的催化性能,结果表明NGO对AP的热分解具有较好的催化效果,NGO的加入能降低AP的起始分解温度,并能显著提高AP的表观分解热。
NC是双基系固体推进剂的粘结剂基体,其热分解提供的能量远大于复合推进剂等推进剂中的粘结剂所提供的能量[14-15]。同时,NC作为双基系固体推进剂的主要组分,其热分解对固体推进剂的能量水平有决定性的作用,提高NC的表观分解热,可以显著提高固体推进剂的能量水平[15]。目前,NGO对固体推进剂中粘结剂热分解的催化性能鲜有报道,研究NGO对其热分解的催化性能具有重要的理论和实际应用价值。基于此,本研究采用溶剂-非溶剂法制备了不同质量比的NGO/NC复合含能材料,利用同步热分析仪(TG-DSC),研究了NGO对NC热分解的催化性能。
2 实验部分
2.1 试剂与仪器
试剂: 硝化纤维素,泸州北方化学工业有限公司; 石墨粉,上海华谊集团华原化工有限公司; 超纯水,自制; 高锰酸钾、浓硫酸、磷酸、过氧化氢、盐酸、无水乙醇、浓硝酸、N,N-二甲基甲酰胺,成都市科龙化工试剂厂; 以上试剂均为分析纯。
仪器: Vario EL Cube型元素分析仪,德国Elementar公司; ESCALAB250型X-射线光电子能谱仪,美国Thermo Scientific公司; Spectrum One型傅里叶变换红外光谱仪,美国PE仪器公司; Ultra55场发射扫描电子显微镜,德国Carl Zeiss光学仪器有限公司; SDT Q600同步热分析仪,美国TA仪器公司。
2.2 NGO/NC复合含能材料的制备
采用改进的Hummers法[16-17]制备GO,然后利用硝硫混酸对其进行硝化,从而制备NGO[13]。采用溶剂-非溶剂法制备不同质量比的NGO/NC复合含能材料。在室温下,将质量比分别为0.5%、0.75%、1%、1.25%和1.5%的NGO和NC在超声波的作用下均匀分散在N,N-二甲基甲酰胺中; 搅拌下,将分散液逐滴加入至超纯水中,析出样品,反复清洗,冷冻干燥,制得NGO质量比为0.5%~1.5%的NGO/NC复合含能材料。另外,按照上述方法对NC进行了处理,以便对照。
3 结果与讨论
3.1 结构分析
对所制备的NGO进行元素分析测试,发现NGO中含N量为1.303%,表明通过硝化反应,N元素被引入到了NGO中。为了验证NGO中N元素以何种形式存在,对NGO进行了X-射线光电子能谱(XPS)测试,其结果如图1所示。由图1a可以看出,在401.4 eV处显示了一个N1s峰,表明硝化反应将N元素引入到了NGO中,其结果与元素分析结果一致。图1b表明,NGO在285.6 eV处和287.4 eV处分别出现了C—N键和C—O—N键的峰,其结果与文献[18-19]吻合。图1c中有两个明显的峰,分别位于401.7 eV处—NO2峰和400.1 eV处还原—NO2峰,其结果与文献[20-22]一致。XPS测试结果表明,N元素在NGO中是以—NO2和—NH2形式存在的。

a. typical XPS survey spectrum of NGO
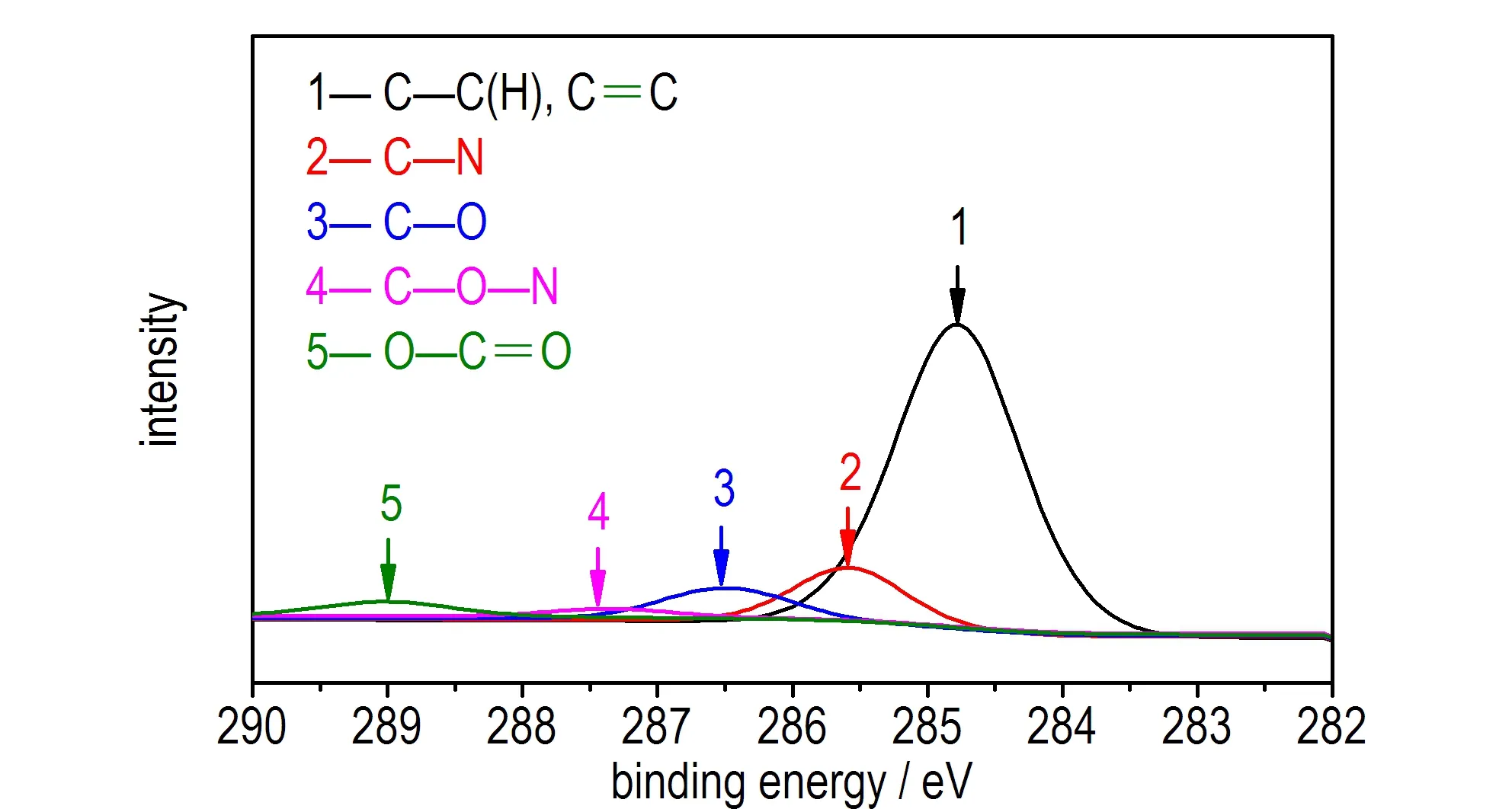
b. C1s XPS spectra
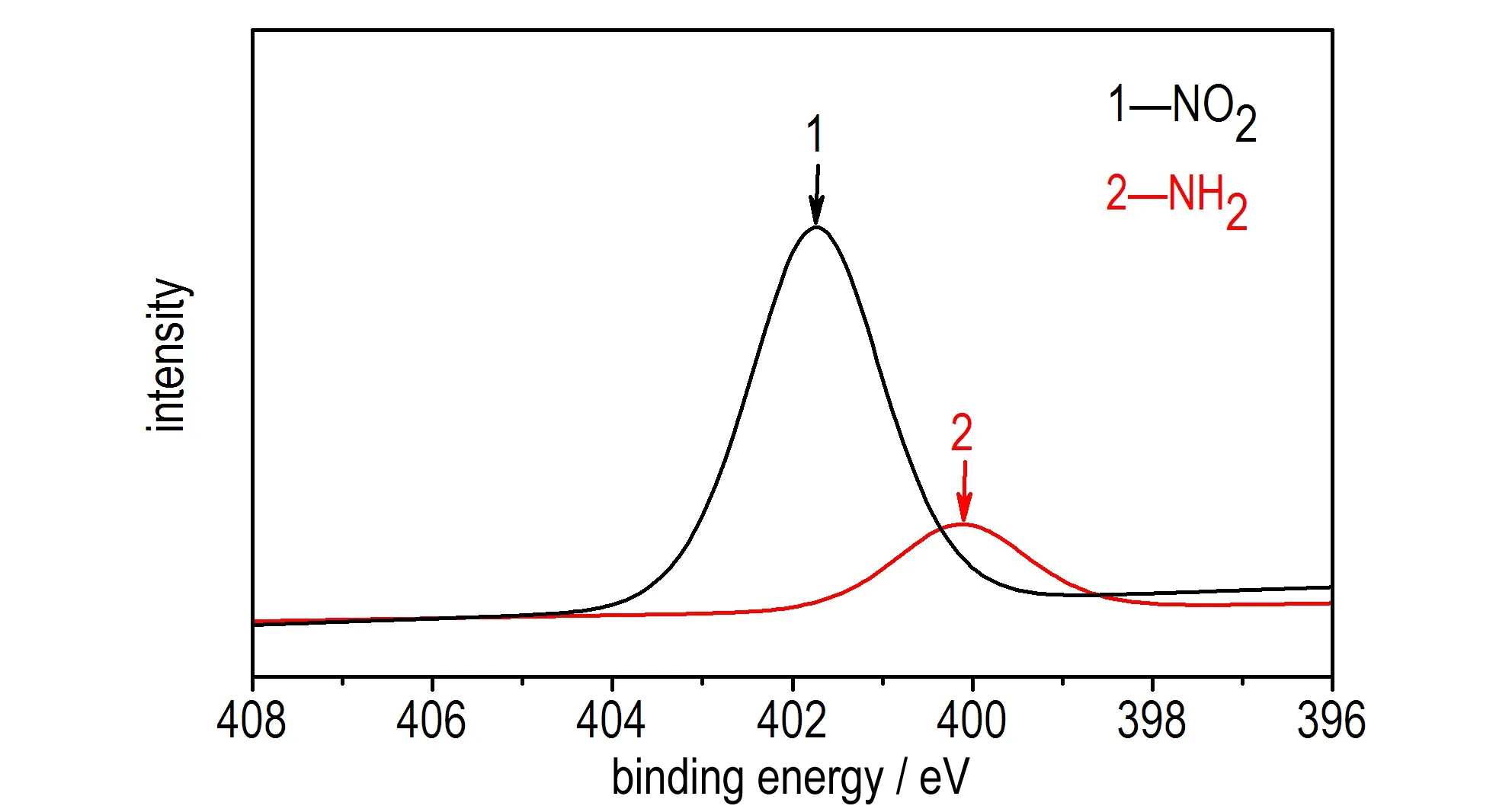
c. N1s XPS spectra
图1 NGO的XPS图谱
Fig.1 XPS spectra of NGO
图2为NGO、NC和1%NGO/NC复合含能材料的傅里叶变换红外光谱(FT-IR)图。由图2可以看出,NGO在3432,2919,2847 cm-1出现了三个特征吸收峰,分别对应的是NGO中—OH或—NH2、—CH2和—CH的伸缩振动峰,且—CH的伸缩振动峰证明了—CH2的存在; 在1727,1056 cm-1出现的两个特征吸收峰分别对应的是羧基中的CO键和环氧基团C—O—C中的C—O键; 在1634 cm-1处的谱峰对应的是—NO2的伸缩振动峰,在1386 cm-1和1224 cm-1处出现的两个伸缩振动峰分别对应的是NGO中C—NO2其测试结果与XPS测试结果一致; 587 cm-1处为C—C—C面内弯曲振动峰。NGO的FT-IR测试表明,NGO中有含能基团和羟基、羧基和环氧基团等亲核含氧基团存在。由NC的FT-IR图谱可以看出,NC在3432,2919,2847 cm-1出现了三个特征吸收峰,分别对应的是—OH、—CH2和—CH的伸缩振动峰; 在1634 cm-1处为—NO2的伸缩振动峰,1258 cm-1为O—NO2键的特征吸收峰; 1457 cm-1为C—O的伸缩振动峰; 1072 cm-1为C—O—C的伸缩振动峰; 在587 cm-1处并没有出现C—C—C面内弯曲振动峰[23]。由1%NGO/NC复合含能材料的FT-IR图谱可以得知,其图谱在1727 cm-1处并没有出现CO键的特征峰; 在2919,2847,1457 cm-1处的特征峰位置较NGO或NC中这三个特征峰的位置没有发生改变; 在1258 cm-1处为O—NO2伸缩振动峰强度相较NGO和NC的都有所减弱,这是由于NGO加入到NC后产生了共轭效应导致的; 在587 cm-1处出现了C—C—C面内弯曲振动峰。NGO、NC和1%NGO/NC复合含能材料的FT-IR测试表明,当1% NGO加入到NC后,不会改变NC的结构。
图2 NGO、NC和1%NGO/NC复合含能材料的傅里叶变换红外光谱图
Fig.2 FT-IR spectra of NGO, NC and 1%NGO/NC energetic composite
3.2 NGO/NC的形貌分析
图3为NGO、NC和1%NGO/NC复合含能材料的场发射扫描电子显微镜(SEM)图,其中插图为各自样品的表观形貌图。由图3a可以看出,NGO的表观形貌为黑色的海绵状,表明NGO的微观形貌为连续的软薄膜状,其表面基本光滑,部分的褶皱是由于缺陷的存在引起的。经溶剂-非溶剂法处理后NC的表观形貌为乳白色的粉末状(图3b),由图3b可以看出,NC的微观形貌呈现出高度多孔的三维网络结构。图3c中插图为1%NGO/NC复合含能材料的表观形貌图,从该图可以其表观形貌为灰色的纤维状,由图3c、图3d可以看出,1%NGO/NC复合含能材料的微观形貌仍为多孔的三维网络结构,与图3a对比发现,1%NGO/NC复合含能材料中没有发现较大的NGO片层,这可能是由于NGO经超声分散,导致其片层大小的改变。形貌分析进一步表明1%NGO/NC复合含能材料中有NGO的存在。

a. NGO
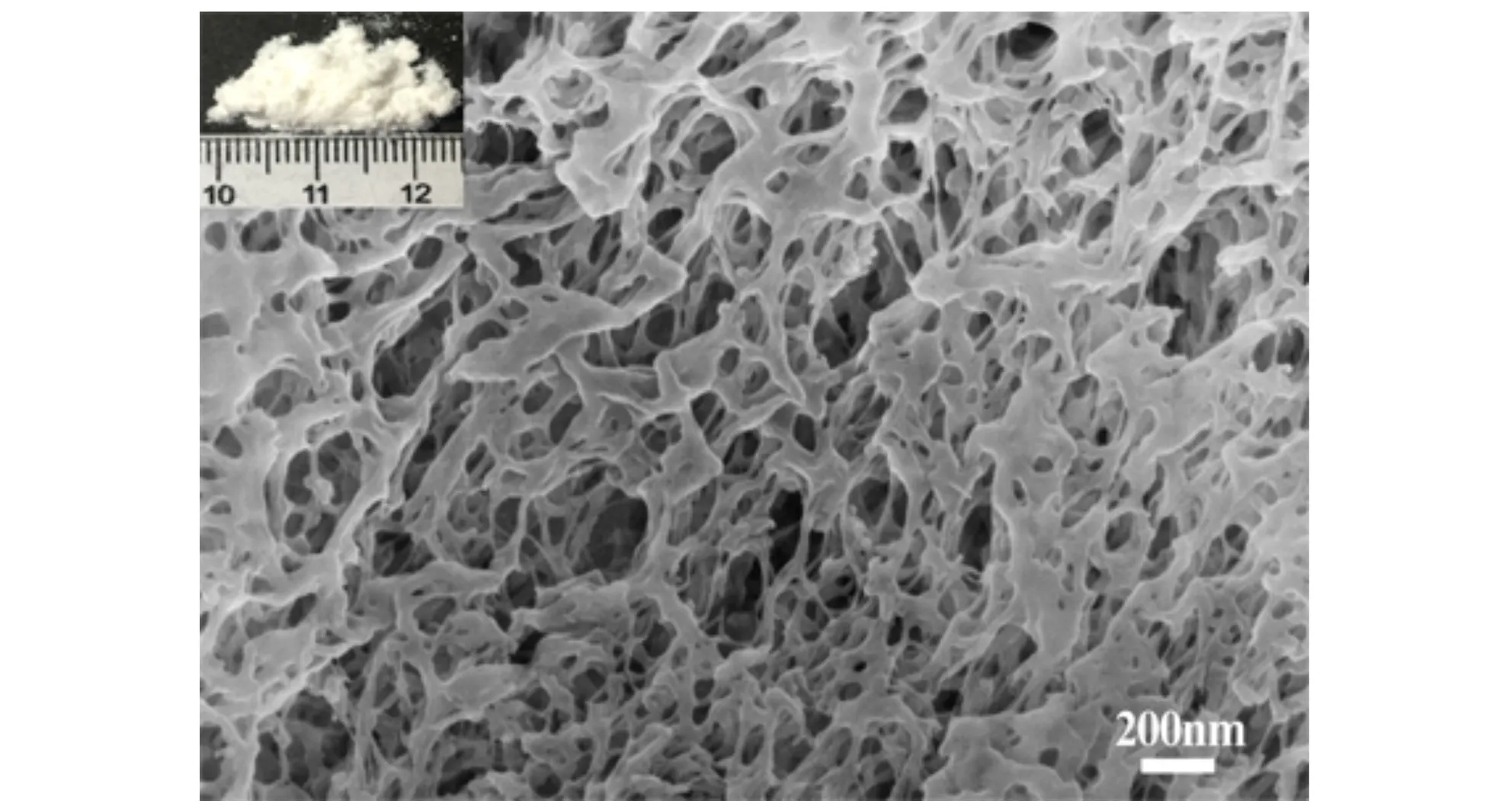
b. NC

c. 1%NGO/NC energetic composite

d. greater magnification of Fig.3c
图3 NGO,NC和1%NGO/NC复合含能材料的扫描电镜图(插图为各自样品的表观形貌图)
Fig.3 SEM images of NGO, NC and 1%NGO/NC energetic composite(insets are the apparent morphology of NGO, NC and 1%NGO/NC energetic composite)
3.3 NGO/NC的热性能分析
为了研究NGO对NC热分解催化性能的影响,采用TG-DSC对NGO、NC、0.5%NGO/NC、0.75%NGO/NC、1%NGO/NC、1.25%NGO/NC和1.5%NGO/NC复合含能材料进行热分析测试(气氛: N2,气体流量: 120 mL·min-1,升温速率: 10 ℃·min-1,温度范围: 室温~500 ℃),其DSC测试结果如图4和表1所示。
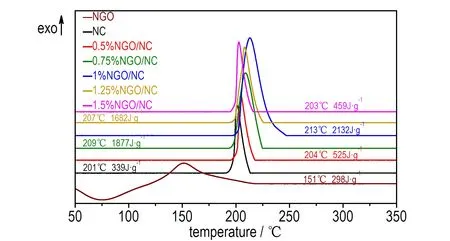
图4 NGO、NC和不同质量比NGO/NC复合含能材料的DSC曲线
Fig.4 DSC curves of NGO, NC and NGO/NC energetic composites in different mass ratios
表1 NGO、NC和不同质量比NGO/NC复合含能材料的放热峰温度和表观分解热
Table 1 Exothermic peak temperature and the apparent decomposition heat of NGO, NC and NGO/NC energetic composites in different mass ratios

sampleTd/℃-ΔH/J·g-1NGO151298NC2013390.5%NGO/NC2045250.75%NGO/NC20918771%NGO/NC21321321.25%NGO/NC20716821.5%NGO/NC203459
Note:Tdis the exothermic peak temperature during the thermal decomposition; -ΔHis the total thermal decomposition heat release of the samples.
由图4可以看出,NGO在75 ℃处有一个由NGO中水蒸发引起的吸热峰,在151 ℃处为其放热峰,其表观分解热为298 J·g-1。NC的热分解过程只在201 ℃处出现一个放热峰,其表观分解热为339 J·g-1。不同质量比NGO/NC复合含能材料的热分解过程也都只出现一个放热阶段,且放热峰面积都明显增强,表观分解热都显著提高(表1); 当NGO添加量从0.5%增加到1%时,复合含能材料的表观分解热随着NGO添加量的增加而提高,NGO对NC的表观分解热有较好的催化效果; 但当NGO添加量超过1%后,复合含能材料的表观分解热随NGO添加量的增加减小; 其中,NGO的添加量为1%时,NGO对NC表观分解热的催化效果最好,1%NGO/NC复合材料的表观分解热为2132 J·g-1,大于NC的339 J·g-1。
引起这种情况可能的原因: NC中O—NO2键能最弱,所以NC热分解的初始反应为O—NO2键断裂生成大量的NO2,初始反应产物进一步分解产生CH2O,然后CH2O与NO2反应,进而放出大量的热(-ΔH=1350 J·g-1),但NO2的生成量不足以使CH2O完全反应[23-26]。NGO的热分解也会产生部分NO2和CH2O。当NGO的添加量不超过1%时,这时NGO热分解提供的NO2能与CH2O充分反应,因此NC的表观分解热会大大增加。当NGO的添加量超过1%后,NC的含量将越来越少,由于NGO含N量的限制,NC热分解提供的NO2和CH2O远大于NGO热分解所提供的。这时,由于复合含能材料中NC含量减少得过多,复合含能材料的热分解过程没有足够的NO2和CH2O使放热反应充分进行,从而导致表观分解热降低。
由图4还可以看出,不同质量比NGO/NC复合含能材料的放热峰温度相对于NC的放热峰温度明显滞后。当NGO的添加量分别为0.5%、0.75%、1%时,NGO/NC复合含能材料的放热峰温度分别为204,209,213 ℃,表明当NGO的添加量小于1%时,NGO/NC复合含能材料的放热峰温度随着NGO添加量的增加滞后得越多,NC的热稳定性越好; 当NGO添加量超过1%后,NGO/NC复合含能材料的放热峰温度随着催化剂添加量的增加逐渐提前,但还是都高于NC的放热峰温度。测试结果表明,NGO的加入减缓了NC的热分解过程,提高了NC热分解的稳定性。
上述现象是由于不同质量比NGO/NC复合含能材料中NC和NGO热分解产生的NO2滞留在NGO/NC复合含能材料的凝聚相骨架中,抑制了NC热分解初始反应的进行,这时将需要更高的温度才能使其反应,所以导致了热分解峰温度的升高[27-28]。随着NGO/NC复合含能材料中NGO质量分数的增加,NC的质量分数相对减少,所产生NO2量也开始减少。当NGO的添加量超过1%后,随着NGO添加量的增加,滞留在NGO/NC复合含能材料凝聚相骨架中的NO2越来越少,NC热分解初始反应将不会被抑制,所以放热峰温度将随之提前。
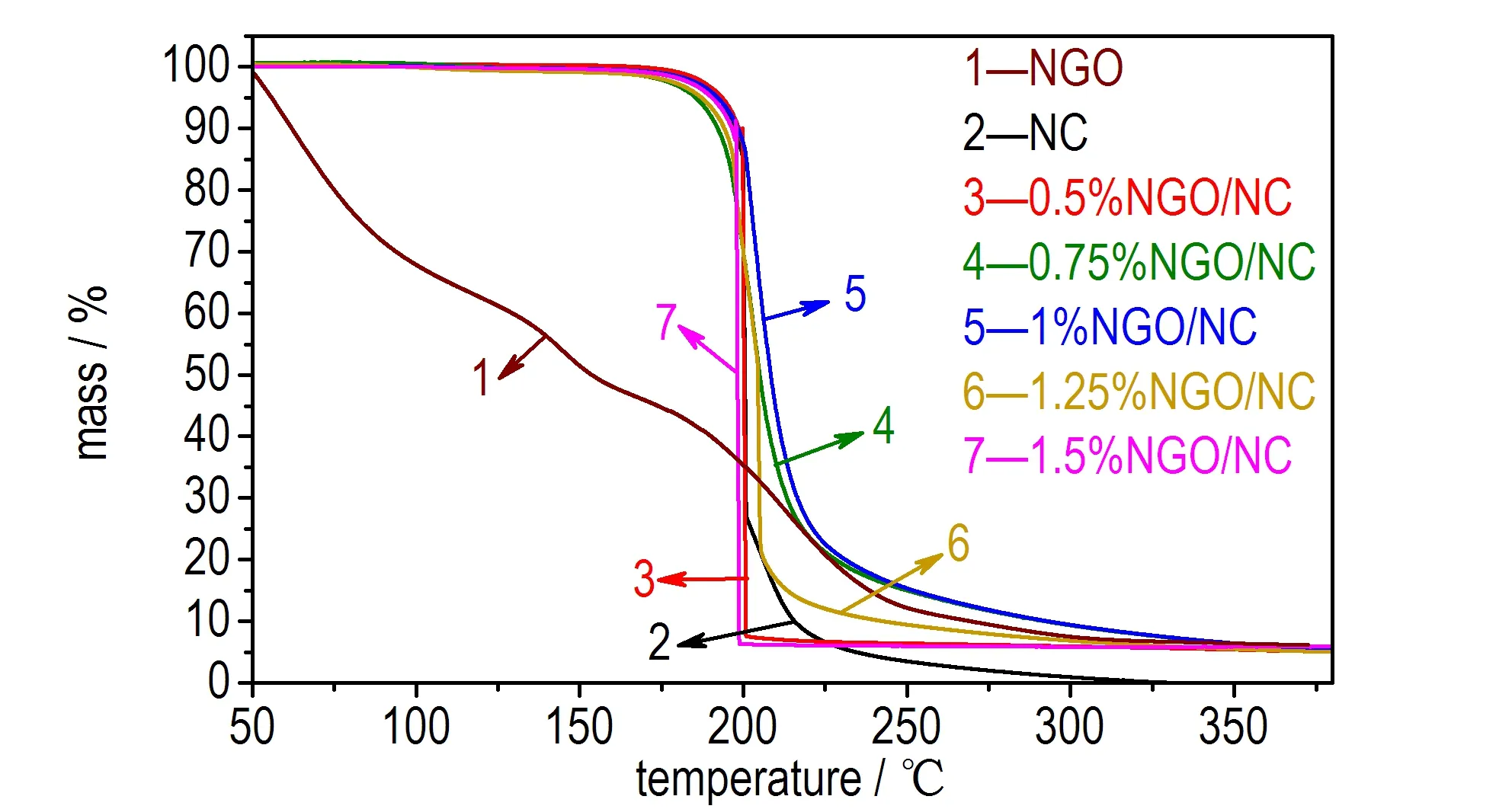
图5 NGO、NC和不同质量比NGO/NC复合含能材料的TG曲线
Fig.5 TG curves of NGO, NC and NGO/NC energetic composites in different mass ratios
图5为NGO、NC和不同质量比NGO/NC复合含能材料的TG曲线。由图5可以发现,当NGO由室温加热至500 ℃时,其质量损失是一个连续的过程,质量损失为96%。NC质量损失为100%,表明其热分解没有固体物质残留在坩埚中。0.5%NGO/NC、0.75%NGO/NC、1%NGO/NC、1.25%NGO/NC和1.5%NGO/NC复合含能材料也都显示一个连续的质量损失过程,质量损失为96%。
4 结 论
(1) 制备了不同质量比NGO/NC复合含能材料,1%NGO/NC复合含能材料为多孔的三维网络结构。
(2) NGO、NC和不同质量比NGO/NC复合含能材料的TG-DSC测试表明,不同添加量的NGO加入都使NC的表观分解热提高,同时也能改善NC的热稳定性,当NGO的添加量为1%时,改善最明显: 复合含能材料的质量损失为96%,表观分解热达到了2132 J·g-1,放热峰温度为213 ℃,相较于NC的表观分解热提高了1739 J·g-1,放热峰温度升高了12 ℃。
参考文献:
[1] 张国涛,周遵宁,张同来,等. 固体推进剂含能催化剂研究进展[J]. 固体火箭技术, 2011, 34(3): 319-323.
ZHANG Guo-tao, ZHOU Zun-ning, ZHANG Tong-lai, et al. Advances on energetic catalysts for solid propellant[J].JournalofSolidRocketTechnology, 2011, 34(3): 319-323.
[2] 王雅乐, 卫芝贤, 康丽. 固体推进剂用燃烧催化剂的研究进展[J]. 含能材料, 2015, 23(1): 89-98.
WANG Ya-le, WEI Zhi-xian, KANG Li. Progress on combustion catalysts of solid propellant[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2015, 23(1): 89-98.
[3] Tong R, Zhao Y, Wang L, et al. Recent research progress in the synthesis and properties of burning rate catalysts based on ferrocene-containing polymers and derivatives[J].JournalofOrganometallicChemistry, 2014, 75(5): 16-32.
[4] 柴玉萍, 张同来. 国内外复合固体推进剂燃速催化剂研究进展[J]. 固体火箭技术, 2007, 30(1): 44-47.
CHAI Yu-ping,ZHANG Tong-lai. Advances on burning rate catalyzer of composite solid propellant at home and abroad[J].JournalofSolidRocketTechnology, 2007, 30(1): 44-47.
[5] 张文文. 硝化石墨烯的制备及其对AP的催化性能研究[D]. 绵阳: 西南科技大学, 2014.
ZHANG Wen-wen.Nitrated graphene oxide and its catalytic activity in thermal decomposition of ammonium perchlorate[D]. Mianyang: Southwest University of Science and Technology, 2014.
[6] Lengelle G, Bizot A, Duterque J, et al.Steady-state burning of homogeneous propellants[J].AmericanInstituteofAeronauticsandAstronautics, 1984, 90(2): 361-407.
[7] Preckel R. Plateau ballistics in nitrocellulose propellants[C]∥Solid Propellant Rocket Conference. Palo Alto, USA, 1964.
[8] Fowler J D, Allen M J, Tung V C, et al. Practical chemical sensors from chemically derived graphene[J].ACSNano, 2009, 3(2): 301-306.
[9] Zhang X, Hikal W M, Zhang Y, et al. Direct laser initiation and improved thermal stability of nitrocellulose/graphene oxide nanocomposites[J].AppliedPhysicsLetters, 2013, 102(14): 141905.
[10] Li R, Wang J, Shen J P, et al. Preparation and characterization of insensitive HMX/graphene oxide composites[J].Propellants,Explosives,Pyrotechnics, 2013, 38(6): 798-804.
[11] 黄海丰, 孟子晖, 周智明, 等. 含能盐和含能离子液体[J]. 化学进展, 2009, 21(1): 152-163.
HUANG Hai-feng, MENG Zi-hui, ZHOU Zhi-ming, et al. Energetic salts and energetic ionic liquids[J].ProgressinChemistry, 2009, 21(1): 152-163.
[12] 黄海丰, 周智明. 基于有机阴离子的含能离子盐研究进展[J]. 火炸药学报, 2012, 35(3): 1-10.
HUANG Hai-feng, ZHOU Zhi-ming. Progress of study on organic aion based energetic salts[J].ChineseJournalofExplosive&Propellants, 2012, 35(3): 1-10.
[13] Zhang W W, Luo Q P, Duan X H, et al. Nitrated graphene oxide and its catalytic activity in thermal decomposition of ammonium perchlorate[J].MaterialsResearchBulletin, 2014, 50(2): 73-78.
[14] 齐晓飞, 严启龙, 刘萌, 等. DIANP对NC溶塑作用的实验与模拟[J]. 含能材料, 2016, 24(3): 269-273.
QI Xiao-fei, YAN Qi-long, LIU Meng, et al. Experiment and simulation on plastication of NC by DIANP[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2016, 24(3): 269-273.
[15] 王泽山. 火炸药科学技术[M]. 第二版. 北京: 北京理工大学出版社, 2002: 272-273.
WANG Ze-shan. The science and technology of explosives[M]. Second Edition. Beijing: Beijing Insitute of Technology Press, 2002: 272-273.
[16] Marcano D C, Kosynkin D V, Berlin J M, et al. Improved synthesis of graphene oxide[J].ACSNano, 2010, 4(8): 4806-4814.
[17] Hummers Jr W S, Offeman R E. Preparation of graphitic oxide[J].JournaloftheAmericanChemicalSociety, 1958, 80(6): 1339.
[18] Mou Z, Chen X, Du Y, et al. Forming mechanism of nitrogen doped graphene prepared by thermal solid-state reaction of graphite oxide and urea[J].AppliedSurfaceScience, 2011, 258(5): 1704-1710.
[19] Ren P G, Yan D X, Ji X, et al. Temperature dependence of graphene oxide reduced by hydrazine hydrate[J].Nanotechnology, 2011, 22(5): 55705-55713.
[20] Bekyarova E, Itkis M E, Ramesh P, et al. Chemical modification of epitaxial graphene: spontaneous grafting of aryl groups[J].JournaloftheAmericanChemicalSociety, 2009, 131(4): 1336-1337.
[21] Adenier A, Cabetdeliry E, Chaussé A, et al. Grafting of nitrophenyl groups on carbon and metallic surfaces without electrochemical induction[J].ChemistryofMaterials, 2005, 17(3): 491-501.
[22] Paula M, Maura B, Mark A, et al. A novel example of X-ray-radiation-induced chemical reduction of an aromatic nitro-group-containing thin film on SiO2to an aromatic amine film[J].Chemphyschem, 2003, 4(8): 884-889.
[23] Makashir P S, Mahajan R R, Agrawal J P. Studies on kinetics and mechanism of initial thermal decomposition of nitrocellulose[J].JournalofThermalAnalysis, 1995, 45(3): 501-509.
[24] Druet L, Asselin M. A review of stability test methods for gun and mortar propellants, II: Stability testing and surveillance[J].JournalofEnergeticMaterials, 1988, 6(3-4): 215-254.
[25] Saunders C W, Taylor L T. A review of the synthesis, chemistry and analysis of nitrocellulose[J].JournalofEnergeticMaterials, 1990, 8(3): 149-203.
[26] Brill T B, Kinloch S A. Condensed phase chemistry of explosives and propellants at high temperature: HMX, RDX and BAMO: discussion[J].PhilosophicalTransactionsoftheRoyalSocietyBBiologicalSciences, 1992, 339(1654): 377-385.
[27] Chen J K, Brill T B. Thermal decomposition of energetic materials 50. Kinetics and mechanism of nitrate ester polymers at high heating rates by SMATCH/FTIR spectroscopy[J].Combustion&Flame, 1991, 85(3): 479-488.
[28] Bergens A, Danielsson R. Decomposition of diphenylamine in nitrocellulose based propellants-I. Optimization of a numerical model to concentration-time data for diphenylamine and its primary degradation products determined by liquid chromatography with dual-amperometric detection[J].Talanta, 1995, 42(2): 171-183.
猜你喜欢
含能材料(2022年8期)2022-08-13
河北果树(2021年4期)2021-12-02
上海公路(2019年3期)2019-11-25
福建基础教育研究(2019年10期)2019-05-28
铜仁学院学报(2018年6期)2018-07-05
厦门理工学院学报(2016年1期)2016-12-01
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
西安建筑科技大学学报(自然科学版)(2014年5期)2014-11-10
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
