
精氨酸侧链与生物小分子间离子氢键强度的快速计算
2017-04-17 08:06:12王长生
辽宁师范大学学报(自然科学版) 2017年1期
王长生, 李 蕾
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
精氨酸侧链与生物小分子间离子氢键强度的快速计算
王长生, 李 蕾
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
提出了1种快速模拟生物分子间离子氢键作用的新方法,并将该方法用于计算模拟带有1个正电荷的精氨酸侧链与甘氨酸二肽分子、碱基尿嘧啶分子、碱基胸腺嘧啶分子等中性分子间的N—H…O=C离子氢键相互作用.计算结果表明:新方法可以快速计算带电精氨酸侧链分子和甘氨酸二肽分子、碱基尿嘧啶分子、碱基胸腺嘧啶分子间形成的N—H…O=C离子氢键的平衡氢键键长和分子间相互作用能;得到的平衡氢键键长与从头计算MP2/6-31+G(d,p)方法得到的平衡氢键键长的绝对偏差均小于0.005 nm;分子间相互作用能与从头计算MP2.5/CBS方法得到的分子间相互作用能的绝对误差均小于8.2 kJ/mol, 相对偏差均小于5.5%; 在同等计算条件下, 新方法的计算效率比从头计算方法快数千倍以上.这些结果表明新方法准确快捷, 在生物大分子体系的分子模拟领域有潜在应用价值.
离子氢键作用;精氨酸;氢键键长;分子间相互作用能;分子模拟
离子氢键作用广泛存在于生物体系中, 在蛋白质-核酸分子识别等过程中起重要作用[1-2], 因此快速准确模拟描述这类氢键作用十分重要.CCSD(T)/CBS方法和MP2.5/CSB方法可以准确计算氢键作用能, 但是由于计算时需要大量的时间和磁盘空间, 因而只适用于小分子体系[3-4].近年来, 本课题组建立了可极化偶极-偶极作用模型, 并将之成功应用于快速预测含有碱基、多肽、酰胺和糖类的N—H…O=C、C—H…O=C、N—H…N和O—H…O型中性氢键复合物的结构和相互作用能[5-8].本文将可极化偶极-偶极作用模型进一步发展, 使之包含计算中性分子与带电分子间相互作用的电荷-偶极作用, 将其应用于计算含有带电精氨酸的N—H…O=C型离子氢键复合物单体间的平衡氢键键长和分子间相互作用能, 并与高精度从头计算方法的计算结果进行比较, 以验证本文方法的合理性.
1 理论模型
在本课题组前期工作[7-9]基础上,提出使用式(1)快速计算生物分子间离子氢键作用强度.

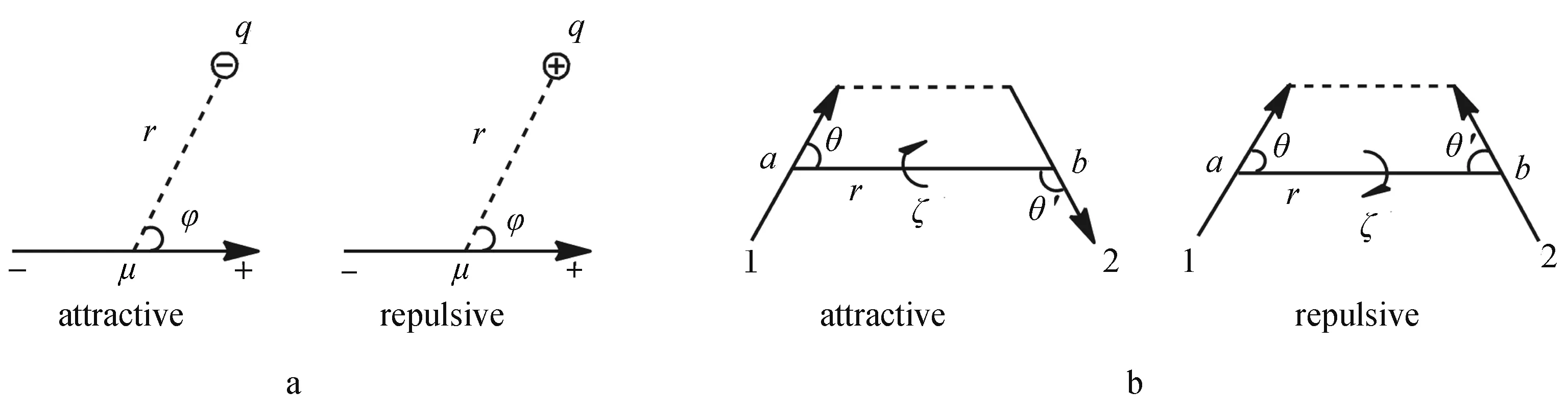
图1 a.点电荷和偶极子间的空间位置关系;b.两个偶极子间的空间位置关系
表1 本文所用参数
Table1Theparametersusedinthiswork
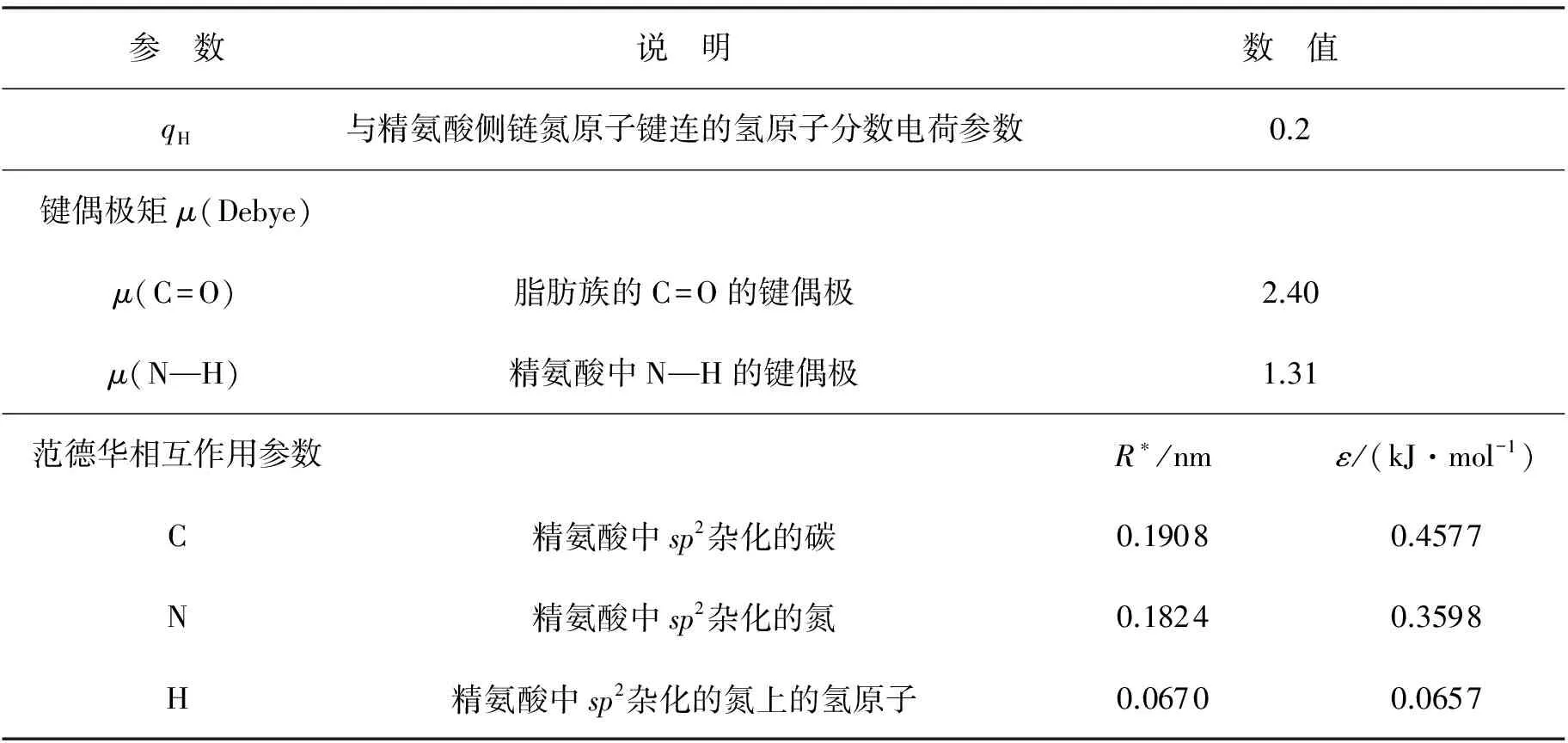
参 数说 明数 值qH与精氨酸侧链氮原子键连的氢原子分数电荷参数0.2键偶极矩μ(Debye)μ(C=O)脂肪族的C=O的键偶极2.40μ(N—H)精氨酸中N—H的键偶极1.31范德华相互作用参数R∗/nmε/(kJ·mol-1)C精氨酸中sp2杂化的碳0.19080.4577N精氨酸中sp2杂化的氮0.18240.3598H精氨酸中sp2杂化的氮上的氢原子0.06700.0657
2 应 用
利用式(1)模型和表1中参数计算了带电精氨酸侧链分子与酰胺分子、甘氨酸二肽分子、碱基尿嘧啶和胸腺嘧啶分子形成的N—H…O=C型离子氢键复合物的结构和作用能, 并与从头计算MP2方法的计算结果进行了比较.图2给出了6个离子氢键复合物单体间平衡氢键键长和相互作用能的计算结果.
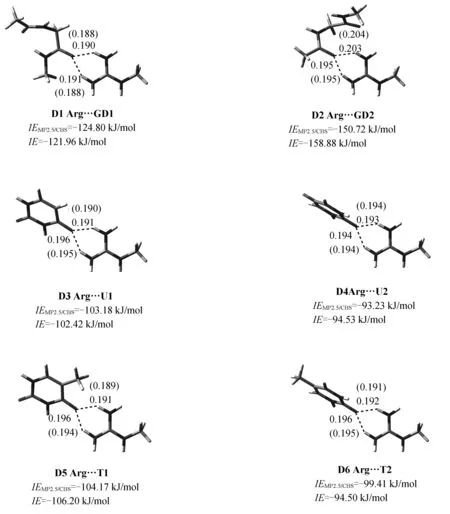
图2 离子氢键复合物的结构和相互作用能
图2中平衡氢键键长的数据表明:本文方法可以较准确地预测N—H…O=C型离子氢键复合物的平衡氢键键长.例如, 对于由带电精氨酸侧链分子(Arg)与甘氨酸二肽分子(GD)形成的离子氢键复合物D1, 从头算MP2/6-31+G(d,p)方法计算得到的2条N—H…O=C型离子氢键平衡键长分别为0.188和0.188nm, 本文方法的计算结果分别为0.190和0.191nm.对于由带电精氨酸侧链分子与碱基尿嘧啶分子(U)形成的离子氢键复合物D4,MP2/6-31+G(d,p)方法计算得到的2条N—H…O=C型离子氢键平衡键长分别为0.194和0.194nm, 本文方法的计算结果分别为0.194和0.193nm.这些结果表明:本文方法可以准确地预测N—H…O=C型离子氢键复合物的平衡氢键键长.对其他离子氢键复合物也可得到同样结论.
图2中相互作用能数据表明本文方法能够准确计算离子氢键复合物的相互作用能.例如, 对于由带电精氨酸侧链分子(Arg)与甘氨酸二肽分子(GD)形成的离子氢键复合物D2,MP2.5/CBS方法给出的相互作用能为-150.72kJ/mol, 本文方法给出的相互作用能为-158.88kJ/mol, 本文方法给出的相互作用能与MP2.5/CBS方法的结果的绝对误差为-8.16kJ/mol, 相对误差仅为5.4%; 对于由带电精氨酸侧链分子(Arg)与碱基尿嘧啶分子(U)形成的离子氢键复合物D4,MP2.5/CBS方法预测的相互作用能为-93.23kJ/mol, 本文方法预测的相互作用能为-94.53kJ/mol, 二者仅相差-1.3kJ/mol, 相对误差仅为1.4%;对于由带电精氨酸侧链分子(Arg)与碱基胸腺嘧啶分子(T)形成的离子氢键复合物D5,MP2.5/CBS方法得到的相互作用能为-104.17kJ/mol, 本文方法得到的相互作用能为-106.20kJ/mol, 二者仅相差-2.03kJ/mol, 相对误差仅为 1.9%.这些结果表明本文方法能够准确计算得到离子氢键复合物的相互作用能.
图3给出了由带电精氨酸侧链分子(Arg)与碱基尿嘧啶分子(U)形成的离子氢键复合物D4和由带电精氨酸侧链分子(Arg)与碱基胸腺嘧啶分子(T)形成的D5的单体间作用能随单体间距离变化的势能曲线.
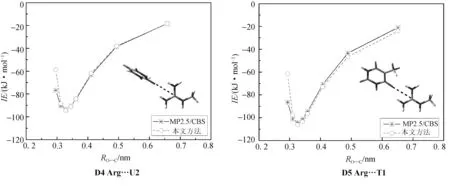
图3 带电精氨酸侧链分子与碱基尿嘧啶、胸腺嘧啶分子间离子氢键作用势能曲线
图3表明: 本文方法计算的势能曲线与高精度从头计算MP2.5/CBS方法的势能曲线符合的很好,本文方法能够较好地描述离子氢键复合物单体间作用强度随分子间距离的变化.
本文方法不但能够较准确地计算离子氢键复合物单体间平衡氢键键长、相互作用能和势能曲线, 而且十分快捷.例如对于离子氢键复合物D4体系, 使用配置为20CPU、64GB内存和9TB硬盘的计算服务器,MP2.5/CBS方法计算相互作用能所需的CPU时间为33h31min24s(120 684s), 本文方法所需的CPU时间仅为27s.本文方法所需时间仅为MP2.5/CBS方法的1/4 470.对于离子氢键复合物D1体系,MP2.5/CBS方法计算相互作用能所需的CPU时间为124h17min10s(447 430s), 本文方法所需的CPU时间仅为34s.本文方法所需时间仅为MP2.5/CBS方法的1/13 160.这些结果说明本文方法十分快捷, 可推广应用于生物大分子体系.
3 结 论
将可极化偶极-偶极作用模型进一步发展, 使之包含计算中性分子与带电分子间相互作用的电荷-偶极作用, 进而提出了一种可快速准确描述生物分子体系中离子氢键作用强度的方法.将本文方法用于带电精氨酸侧链分子和其他中性生物小分子形成的离子氢键复合物体系, 计算结果表明本文方法简洁快速, 可以较准确预测N—H…O=C型离子氢键的平衡氢键键长、离子氢键复合物单体间的相互作用能以及单体间离子氢键作用强度随距离的改变.本文方法在生物大分子体系的分子模拟方面有潜在应用价值.
[1] MEOT-NER M.Update 1 of:strong ionic hydrogen bonds[J].Chem Rev,2012,112(1):22-103.
[2] 李蕾,黄翠英,姜笑楠,等.精氨酸侧链和核酸碱基间离子氢键作用强度分析[J].高等学校化学学报,2016,37(8):1460-1467.
[5] GAO X C,HUANG C Y,WANG C S.Rapid evaluation of the interaction energies for hydrogen-bonded uracil and thymine dimers, trimers and tetramers[J].Comput Theor Chem,2014,1048:46-53.
[6] 王长生,黄翠英.核酸碱基间氢键距离和作用能的快速预测[J].辽宁师范大学学报(自然科学版),2016,39(3):368-372.
[7] LI S S,HUANG C Y,HAO J J,et al.A polarizable dipole-dipole interaction model for evaluation of the interaction energies for N-H…O=C and C-H…O=C hydrogen-bonded complexes[J].J Comput Chem,2014,35(6):415-426.
[8] HAO J J,WANG C S.Rapid evaluation of the interaction energies for carbohydrate-containing hydrogen-bonded complexes via the polarizable dipole-dipole interaction model combined with NBO charge or AM1 charge[J].RSC Adv,2015,5(9):6452-6461.
[9] 王嘉,王长生.特殊氢原子与丙氨酸二肽构象稳定性的理论研究[J].辽宁师范大学学报(自然科学版),2004,27(3):320-323.
Rapid evaluation of the ionic hydrogen bonding strength between charged arginine side chain and small biomolecule
WANGChangsheng,LILei
(School of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, China)
A new scheme is proposed in this paper to accurately and efficiently evaluate the ionic hydrogen bonding interaction between biomolecules. The scheme is estimated by applying to a series of complexes containing an arginine side chain molecule and a small biomolecules such as glycine dipeptide molecule, nucleic acid base uracil molecule, nucleic acid thymine molecule, etc. The calculated results shows that our method not only produces the equilibrium hydrogen bond lengths as accurate as those produced by the MP2/6-31+G(d,p) method, the intermolecular interaction energies as accurate as those produced by the high quality MP2.5/CBS method, but very efficient as well, demonstrating the scheme proposed in this paper is useful in the context of molecular modeling of large biomolecular systems.
ionic hydrogen bonding;arginine;hydrogen bond length;intermolecular interaction energy;molecular modeling
2016-10-20 基金项目:国家自然科学基金资助项目(21173109;21573098) 作者简介:王长生(1963-),男,辽宁大连人,辽宁师范大学教授,博士,博士生导师.
1000-1735(2017)01-0047-05
10.11679/lsxblk2017010047
O641.121
A
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29 03:07:02
核农学报(2020年2期)2020-03-11 08:33:16
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
世界农药(2019年2期)2019-07-13 05:55:12
菏泽学院学报(2017年2期)2017-05-16 08:59:11
粘接(2017年4期)2017-04-25 08:37:20
陕西师范大学学报(自然科学版)(2015年1期)2016-01-16 03:23:30
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
