
基于异质网络的长非编码RNA和蛋白质相互作用的预测算法研究
2017-04-14 01:00郑肖雄朱文琰
计算机应用与软件 2017年3期
郑肖雄 朱文琰
(复旦大学计算机科学技术学院智能信息处理重点实验室 上海 200433)
基于异质网络的长非编码RNA和蛋白质相互作用的预测算法研究
郑肖雄 朱文琰
(复旦大学计算机科学技术学院智能信息处理重点实验室 上海 200433)
长非编码RNA在生物过程中扮演着非常重要的角色,长非编码RNA可以与多种蛋白质结合发挥其生物功能,预测长非编码RNA和蛋白质的相互作用也成为了研究长非编码RNA功能的途径之一。由于长非编码RNA的低保守性,通过提取特征和用机器学习算法预测它和蛋白质之间的相互作用将会不太合适。LPHeteSim算法是一种基于对称路径随机游走的方法,它可以衡量异质长非编码RNA和蛋白质相互作用网络中两者的相关性。在导质网络中,LPHeteSim算法可以有效地预测两者的相互作用,实验结果验证了算法的有效性。
长非编码RNA和蛋白质相互作用 随机游走 异质网络
0 引 言
长非编码RNA(Long Noncoding RNA) 是指一类长度大于200个核苷酸、不编码蛋白质的非编码RNA[1]。在人类转录物组中,只有小部分(约1%)的RNA编码蛋白质,其他的RNA都是不编码蛋白质的非编码RNA,其中的大多数属于长非编码RNA[2]。越来越多的证据表明长非编码RNA在多种层面上(表观遗传调控、转录调控以及转录后调控等)调控基因的表达水平[3]。例如,长非编码RNA参与了X染色体沉默[4],基因组印记以及染色质修饰、转录激活、转录干扰和核内运输等多种重要的生物过程。此外,长非编码RNA还参与了很多疾病的形成,Yang等人[13]从网络的角度系统的研究了长非编码RNA和疾病的关系,发现很多长非编码RNA都和疾病都有联系。长非编码RNA功能的多样性和复杂性是由于和多个蛋白质相互作用[5],它会通过与蛋白质结合实现自己的功能,从而对多个细胞过程进行调控。虽然近年来关于长非编码RNA的研究进展迅猛,但是绝大部分的长非编码RNA的功能仍然是不清楚的。
近年来,由于交联免疫共沉淀生物技术和高通量测序技术的发展,越来越多关于长非编码RNA和蛋白质的相互作用被发现。然而,通过实验发现两者相互作用仍然占很小比例。越来越多研究者通过计算的方法预测长非编码RNA和蛋白质相互作用。2011年,Bellucci等人[6]提出了CatRAPID方法,他们在Xist网络中预测RNA和蛋白质交互。同年,Pancaldi等人[7]用随机森林和支持向量机的机器学习算法,从RNA和蛋白质的物理特性,二级结构等抽取特征,预测RNA和蛋白质的交互。Muppirala等人[8]从蛋白质和RNA序列中抽取特征,使用同样的算法预测长非编码RNA和蛋白质的相互作用。2013年,Wang等人[9]用朴素贝叶斯和扩展的朴素贝叶斯分类器进行预测。同年,Lu等人[10]提出了lncPro算法,他们将RNA和蛋白质编码成数字向量,再用矩阵乘法对RNA-蛋白质对打分的方式进行预测。
以上算法都是基于RNA和蛋白质自身的特性预测两者的相互作用,然而,由于长非编码RNA的低保守性[11],基于长非编码RNA本身的特性预测长非编码RNA和蛋白质的相互作用可能会遇到很多困难。一些研究者采用了通过相互作用网络的拓扑结构预测两者的相互作用,他们通过构建长非编码RNA和蛋白质的相互作用网络,用网络中的隐含信息预测新的交互。2015年Li等人[12]用异种网络随机游走方法预测长非编码RNA和蛋白质交互。他们用长非编码RNA的表达特征相似性构建长非编码RNA相似度网络、STRING数据库构建蛋白质相互作用网路和NPInter数据库构建蛋白质相互作用网络,并提出了LPIHN方法,一种在异质网络进行重启型随机游走的策略,取得了很好的效果。同年,Yang等人[13]采取了基于路径的双向随机游走的算法(HeteSim),预测长非编码RNA和蛋白质相互作用。然而他们在计算蛋白质之间的相似度时,并没有考虑到蛋白质之间相似度的强弱关系,本文将在此基础上采用基于HeteSim的方法,并通过融合不同的蛋白质相似度网络,构造出更加可靠的蛋白质相似度网络,预测长非编码RNA和蛋白质的相互作用。
1 基于LPHeteSim算法预测
1.1 HeteSim 算法

异质网络中不同的结点之间的边都包含着不同的信息,通过不同的相关路径得到的相似度,其背后隐含的意义是各不相同的。例如,在异质长非编码RNA和蛋白质相互作用网络中,长非编码RNA可以通过“长非编码RNA-蛋白质-蛋白质”(LPP)和“长非编码RNA-长非编码RNA-蛋白质”(LLP)两种路径连接蛋白质。然而,这两种路径背后表达的意义是不一样的,前者联合了长非编码RNA-蛋白质相互作用网络和蛋白质-蛋白质相似度网络,后者联合了长非编码RNA-长非编码RNA相似度网络和长非编码RNA-蛋白质相互作用网络。LPP是通过利用蛋白质相似度网络得到新的长非编码RNA和蛋白质相互作用,LLP则是利用了长非编码RNA相似度网络得到新的长非编码RNA和蛋白质相互作用。
由于异质网络中的路径含有隐藏语义的,这使得对象之间的关系依赖于给定的相关路径,HeteSim就是在此思想下提出来的。
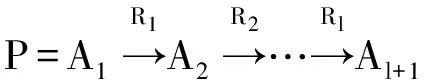
HeteSim(o1,ol|R1∘R2∘…∘Rl-1∘Rt)=
HeteSim(Oi(o1|R1),Ij(ol|Rl)|R2∘…∘Rl-1)
(1)
其中,O(o1|R1)是对象o1基于关系R1的出度邻居集合,O(ol|Rl)对象ol基于关系Rl的入度邻居集合。Oi(o1|R1)/Ij(ol|Rl)是O(o1|R1)/O(ol|Rl)中的i/j个对象。
在HeteSim中,如果对象o1和ol在关系I中相同,称之为自我相关的,本文将定义这种关系为自相关关系(I关系)。
定义2(自相关HeteSim)s与t是相同类型时,基于自相关关系下做相似性度量时,其相似度分数定义如下:
HeteSim(s,t|I)=δ(s,t)
(2)
如果s和t相同时,δ(s,ol)=1,否则δ(s,t)=0。
1.2LPHeteSim算法
原始的自相关HeteSim分数定义s与t相同时δ(s,t)=1,显然,此种定义不适合长非编码RNA-蛋白质异质网络,在Yang等人的论文中对其进行了如下改进:
定义3 如果源对象o1和目标对象ol有相互作用时,在其自相关关系I下做相似性度量时,自相关关系分数定义如下:
HeteSim(o1,ol|I)=δ(o1,ol)
(3)
如果x和y有相互作用,δ(o1,ol)=1,否则δ(o1,ol)=0。
然而,在衡量蛋白质之间的相似性时,直接将其相似度归为1或者0将影响预测结果的准确性。实际上,现在有很多生物数据库都提供蛋白质与蛋白质之间的相似度,他们通过生物实验、计算方法对其相似度进行打分,例如STRING数据库等,本文将直接采用这些数据库的打分。所以,本文对HeteSim进行了改进,提出了LPHeteSim算法,该算法的自相关性定义如下:
定义4 源对象是长非编码RNA(l),目标对象时蛋白质(p),其自相关关系分数定义如下:
LPHeteSim(l,p|I)=δ(l,p)
(4)
如果l和p有相互作用时,δ(l,p)=1,否则δ(l,p)=0。
源对象是蛋白质(p1),目标对象是蛋白质(p2),其自相关关系分数定义如下:
LPHeteSim(p1,p2|I)=δ(p1,p2)
(5)
δ(p1,p2)=p(p1,p2)
(6)
其中,p(p1,p2)是蛋白质p1和蛋白质p2的相似度。
对于基于路径的双向随机游走方法,计算两个结点的相似度,最重要的是路径选择,在Yang的论文中采用的“长非编码RNA-蛋白质-蛋白质”(LPP)路径要明显好于其他路径,在此,本文也将会采用LPP路径验证LPHeteSim算法的有效性。
2 基于LPHeteSim-SNF算法的预测
2.1 相似度网络融合算法
在计算蛋白质之间的相似度时,有多种计算方式,例如计算蛋白质之间的基因本体(GeneOntology)相似度、域相似度和序列相似度等等,由此,构成不同的蛋白质相似度矩阵。然而,不同的计算方式得到的相似度都有可能含有噪音,最好的方式是通过融合多个相似度矩阵,取每个相似度网络中的可靠的相似度,去除相似度网络中不可靠的相似度,即噪音。在此本文整合四种计算蛋白质相似度的方法,对不同度量方式的蛋白质矩阵进行融合,得到一个更加稳健的相似度矩阵。本文采用了由Wang等人[15]在2014年提出的相似网络融合算法。
假设有n种蛋白质和m种度量蛋白质相似度的方式。蛋白质相似网络记作图G=(V,E),V代表蛋白质集合{x1,x2,…,xn},即顶点,E代表蛋白质之间的权重,即边,相似度矩阵记为W,W(xi,xj)代表蛋白质xi和蛋白质xj之间的相似度。
为了计算多个度量下的蛋白质相似度融合矩阵在顶点集V上定义一个全稀疏核P=D-1W,D是一个对角矩阵,由蛋白质相似度矩阵W得到,并对其进行正则化,D(i,i)=∑jW(i,j),所以∑jP(i,j)=1。然而,正则化之后可能会引入数值不稳定性,因为它涉及到W的对角项上的自我相似性,更好的一种正则化形式如下所示:
(7)
用Ni表示xi在图G中的邻居结点,在图G中,用KNN计算其局部相关性,设置非邻居结点值为0。假设邻居结点的相似性比非邻居结点的相似性可靠,之后再通过网络将相似性传播给其他结点:
(8)
在矩阵P中包含着相似性网络的全部信息,矩阵S中携带着相对重要的信息。这里将把矩阵P作为初始状态,然后对其进行迭代,矩阵S将作为融合过程中的核矩阵,用来获得矩阵的局部结构性和增强计算效率。

(9)
(10)

(11)
因为矩阵S是P的KNN图,所以此迭代的过程中将慢慢降低矩阵中噪音的影响。从迭代步骤中看出,相似度仅会通过结点之间共同的邻居传播,这会让相似度矩阵更加稳固。而且不同度量下的相似度矩阵都会从其他度量下的相似度矩阵得到信息补充,这样就达到了相互融合的目的。对于m>2时,上述的迭代步骤将演变成:
(12)
2.2LPHeteSim-SNF算法
本文采用了四种度量蛋白质之间相似度的数据集,其中三种是通过蛋白质本身的特性对其相似度打分(基因本体相似性、域相似性、序列相似性),第四种是采用公共数据库,即String数据库中的蛋白质相似度分数。
1) 基于基因本体的蛋白质相似度计算
基因本体[16]是一个用规范化的基因和基因产物特性的术语描绘或词义解释的数据库,其中的术语主要涉及到生物学的三个方面:细胞组件、分子功能和生物过程。基因本体是一个有向无环图,它包含三种关系:分别是is_a、part_of和regulates。为了衡量蛋白质pi和蛋白质pj的基因本体相似度,这里采用Jaccard值[17]计算它们之间的相似度。蛋白质pi对应的基因本体术语集合为ti,蛋白质pj对应的基因本体术语集合为tj。则两者的相似度如下所示:
(13)
其中,分子是两个蛋白质对应的基因本体集合的交集,分母是两者的并集。
2) 基于蛋白质域的相似度计算
蛋白质通常由一个或多个功能区域组成,被称为蛋白质域。结构域的不同组合方式尝试的蛋白质也不尽相同。Pfam数据库[18]中搜集了大量的蛋白质家族,它依赖于由多序列比对和隐马尔科夫模型产生的结果。
具有相同域的蛋白质之间的相似度会很大,可以将每个蛋白质表示为一个二值向量,如果蛋白质中存在这个域,则向量中对应该域的值为1,否则为0。最后,用广义的Jaccard值计算它们之间的相似度。
(14)
其中,‖·‖是向量的模,xi和xj分别对应蛋白质pi和蛋白质pj的域向量,SomainSim(pi,pj)为二者的蛋白质域相似度。
3) 基于蛋白质序列的相似度计算
同样通过序列的方式计算蛋白质之间的相似度,蛋白质序列数据可以从UniProt数据库[19]中获得。Uniprot数据库是一个可供免费使用的蛋白质序列与功能信息数据库,它包含了大量蛋白质的详细信息。
本文采用规范化过的Smith-Waterman方法[20]计算蛋白质之间的序列相似度。
(15)
其中sw(pi,pj)是蛋白质pi和蛋白质pj的Smith-Waterman得分。为了将规范化之后的蛋白质相似度得分应用于所有的蛋白质对中,将计算之后的得分平均化:
(16)
为此,由上式可以得到蛋白质pi和蛋白质pj之间的序列相似度。
4) 基于STRING的蛋白质相似度计算
STRING数据库[21]是一个包含大量蛋白质相互作用的数据库,它覆盖了超过2 000多种生物,不仅整合了已被实验验证的蛋白质相互作用,还包括通过计算方法得到的蛋白质相互作用。
在由STRING数据库中得到的得分中,最高分数为999,为了将得分控制在[0,1]内,对其进行如下操作:
(17)
因此,可以得到蛋白质pi和蛋白质pj之间的STRING相似度。
在得到四种蛋白质相似度网络,用相似度网络融合算法(SNF)进行融合。在联合长非编码RNA和蛋白质相互作用网络,构建异质长非编码RNA和蛋白质相互作用网络。在此异质网络中,执行LPHeteSim算法,预测新的长非编码RNA和蛋白质相互作用。
3 实验分析
本文实验从NPInter(V2.0)数据库[22]中抽取所有非编码RNA和蛋白质相互作用的数据,再从NONCODE(V4.0)数据库[23]中抽取人类的长非编码RNA数据,如果交互数据中的非编码RNA是长非编码RNA,抽取它与蛋白质的交互数据。因此,可以构建出长非编码RNA和蛋白质相互作用网络。对于在长非编码RNA和蛋白质相互作用网络中的蛋白质,分别从GO数据库、Uniprot数据库、Pfam数据库中获得它们的GO信息、序列信息和域信息,再分别计算出蛋白质之间的GO相似度、序列相似度以及域相似度。本文同样从STRING(v10.0)数据库中获取由STRING数据库计算的蛋白质之间相互作用的得分,并计算出蛋白质之间的STRING相似度得分。为此,经过上述操作,可以获得四种蛋白质相似度网络。同样对数据集进行了预处理,去除只和一个蛋白质相互作用的长非编码RNA,因为无法采用留一交叉验证进行验证。同样丢弃和其他所有蛋白质的相似度都为0的蛋白质,因为,它们只会给实验带来噪音。最后,将得到的长非编码RNA的数量、蛋白质的数量以及两者的相互作用数量统计如表1所示。
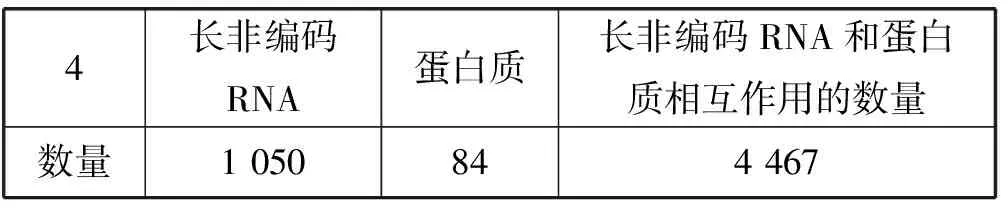
表1 长非编码RNA和蛋白质以及它们相互作用的数量
本文采用留一交叉验证验证我们的算法有效性。对于每种实验结果,分别得到它们的ROC曲线以及ROC曲线下的面积(AUC)值衡量算法的有效性。实验中所采用的正样本即是已知的长非编码RNA和蛋白质相互作用,负样本即是未被报道的长非编码RNA和蛋白质相互作用。
先基于四个蛋白质相似度网络测试LPHeteSim算法,对比算法为Yang等人所采用的HeteSim算法。
LPHeteSim和HeteSim算法在基因本体(Go)、蛋白质域、蛋白质序列、STRING数据库四个数据集上ROC曲线如图1、图2、图3和图4所示。实线代表LPHeteSim算法在基于此蛋白质相似度网络的ROC曲线,虚线代表HeteSim算法的ROC曲线。LPHeteSim的ROC曲线显然比HeteSim的高,为此可以说明算法改进的有效性。然而,两者都会在其中一点汇聚,因为LPHeteSim可以提高预测的精确度,并不会增多预测数量。
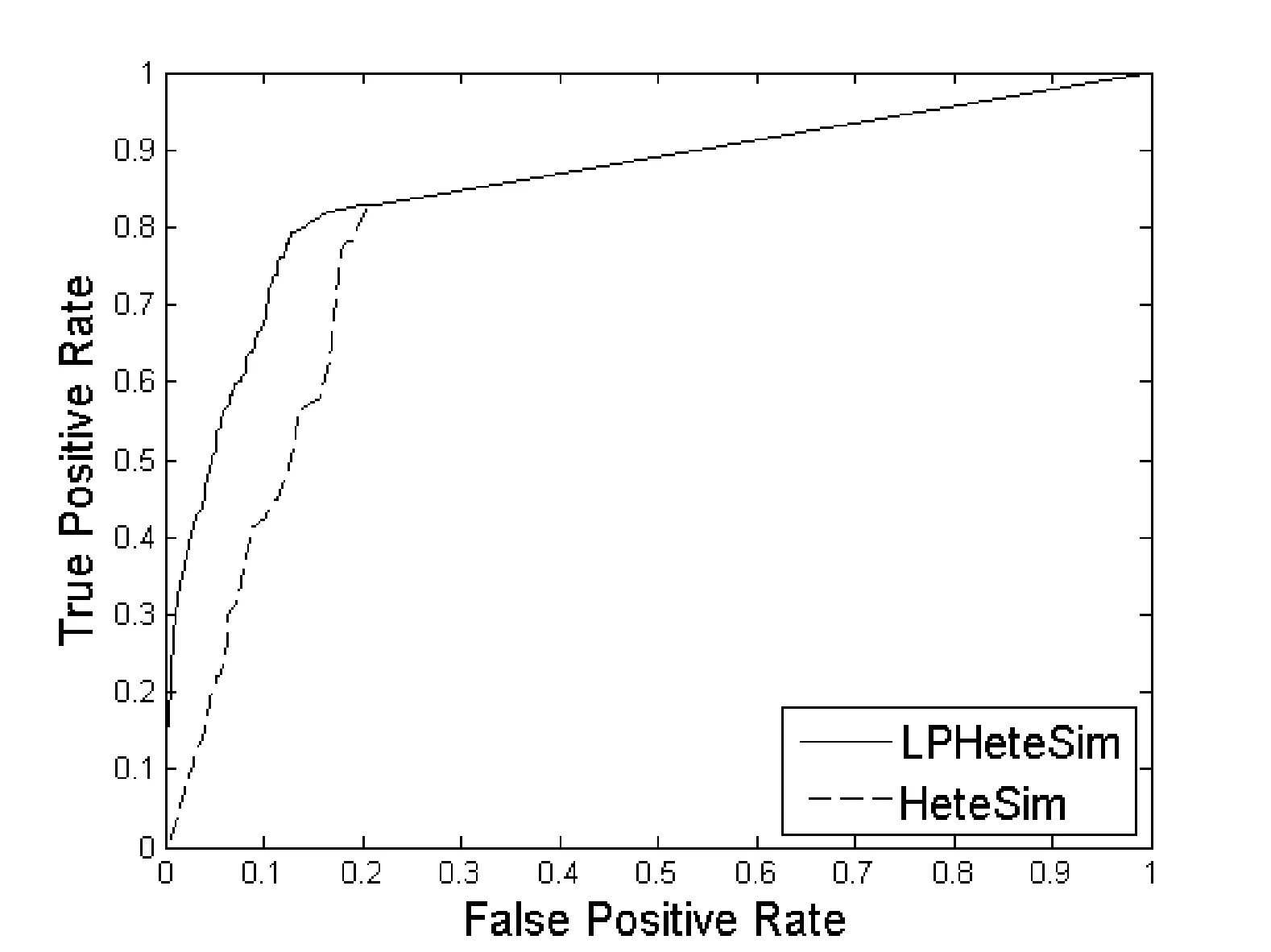
图1 基于蛋白质基因本体相似度的LPHeteSim和HeteSim的ROC曲线

图2 基于蛋白质域的LPHeteSim和HeteSim的ROC曲线
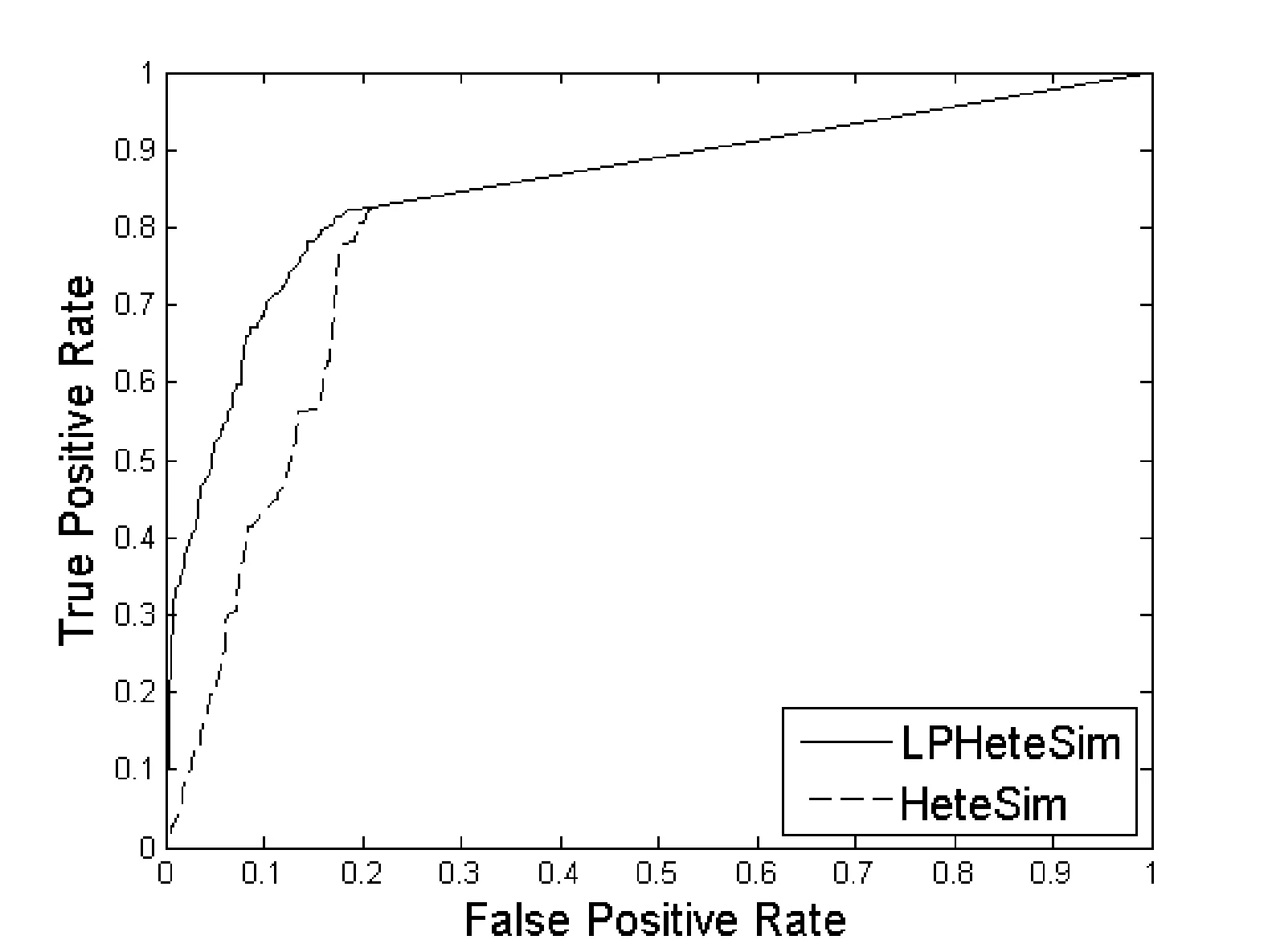
图3 基于蛋白质序列相似度的LPHeteSim和HeteSim的ROC曲线

图4 基于STRING数据库的LPHeteSim和HeteSim的ROC曲线
LPHeteSim和HeteSim算法的AUC值如表2所示, LPHeteSim算法在各个蛋白质相似度网络上的表现均要比HeteSim算法好很多,它们的AUC值分别为0.858 4、0.848 9、0.856 5和0.797 2。均要比HeteSim算法的预测效果好,为此,验证了本文的算法有效性。

表2 LPHeteSim和HeteSim在各个数据集上的AUC值
然后用SNF算法融合四个蛋白质相似度网络,并在基于融合之后的相似度网络上执行LPHeteSim算法,预测新的长非编码RNA和蛋白质相互作用。预测结果的ROC曲线如图5所示。
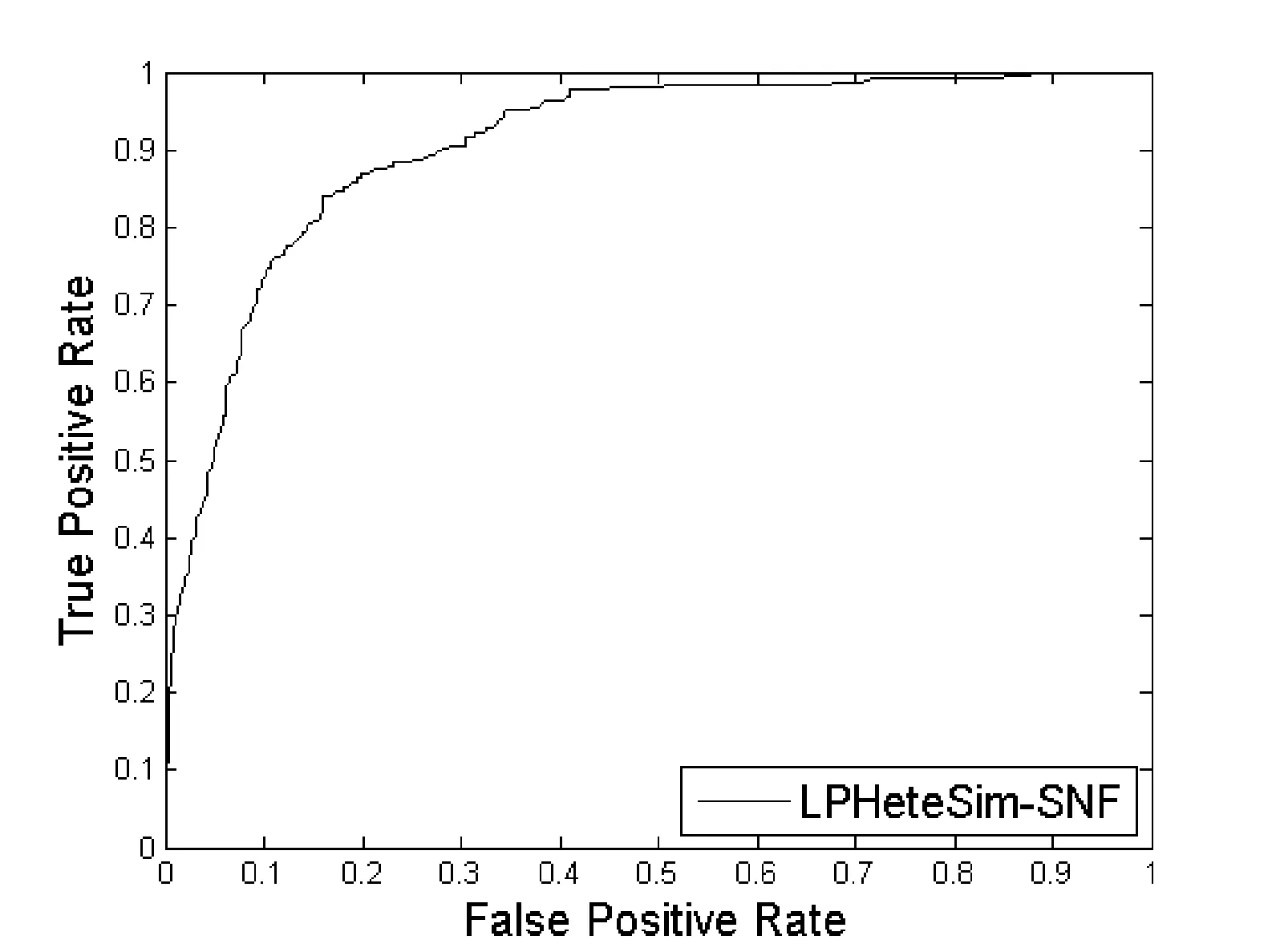
图5 基于融合蛋白质相似度网络的LPHeteSim的ROC曲线
可以从ROC曲线和AUC值看出,融合之后的AUC值为0.906 8,明显比在每个蛋白质相似度网络的预测效果高。因此,相似度网络融合算法可以减少每个矩阵中的噪音,可以有效地提高预测算法的准确性。
4 结 语
本文在HeteSim算法的基础上,通过改进自相关关系相似度的计算方式,提出了LPHeteSim算法。在单个蛋白质相似度网络上的实验表明,改进之后的算法的有效性要好于未改进的HeteSim算法。
为了进一步提高预测准确性,本文采用融合蛋白质相似度网络的方法,构造出置信度更高的蛋白质相似度矩阵。实验结果表明,在融合之后的蛋白质相似度网络执行预测算法,要好于在单个蛋白质相似度网络上的预测结果。因此,结合相似度融合算法和LPHeteSim算法,可以有效地提高预测新的长非编码RNA和蛋白质相互作用的有效性。
[1] Bonasio R,Shiekhattar R.Regulation of transcription by long noncoding RNAs[J].Annual Review of Genetics,2014,48:433.
[2] International Human Genome Sequencing Consortium.Finishing the euchromatic sequence of the human genome[J].Nature,2004,431(7011):931-945.
[3] Geisler S,Coller J.RNA in unexpected places:long non-coding RNA functions in diverse cellular contexts[J].Nature reviews Molecular cell biology,2013,14(11):699-712.
[4] Engreitz J M,Pandya-Jones A,McDonel P,et al.The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome[J].Science,2013,341(6147):1237973.
[5] Zhu J J,Fu H J,Wu Y G,et al.Function of lncRNAs and approaches to lncRNA-protein interactions[J].Science China Life Sciences,2013,56(10):876-885.
[6] Bellucci M,Agostini F,Masin M,et al.Predicting protein associations with long noncoding RNAs[J].Nature Methods,2011,8(6):444-445.
[7] Pancaldi V,Bähler J.In silico characterization and prediction of global protein-mRNA interactions in yeast[J].Nucleic Acids Research,2011:gkr160.
[8] Muppirala U K,Honavar V G,Dobbs D.Predicting RNA-protein interactions using only sequence information[J].BMC Bioinformatics,2011,12(1):1.
[9] Wang Y,Chen X,Liu Z P,et al.De novo prediction of RNA-protein interactions from sequence information[J].Molecular BioSystems,2013,9(1):133-142.
[10] Lu Q,Ren S,Lu M,et al.Computational prediction of associations between long non-coding RNAs and proteins[J].BMC Genomics,2013,14(1):1.
[11] Pang K C,Frith M C,Mattick J S.Rapid evolution of noncoding RNAs:lack of conservation does not mean lack of function[J].Trends in Genetics,2006,22(1):1-5.
[12] Li A,Ge M,Zhang Y,et al.Predicting Long Noncoding RNA and Protein Interactions Using Heterogeneous Network Model[J].BioMed Research International,2015,2015.
[13] Yang J,Li A,Ge M,et al.Prediction of interactions between lncRNA and protein by using relevance search in a heterogeneous lncRNA-protein network[C]//Control Conference (CCC),2015 34th Chinese.IEEE,2015:8540-8544.
[14] Shi C,Kong X,Huang Y,et al.Hetesim:A general framework for relevance measure in heterogeneous networks[J].Knowledge and Data Engineering,IEEE Transactions on,2014,26(10):2479-2492.
[15] Wang B,Mezlini A M,Demir F,et al.Similarity network fusion for aggregating data types on a genomic scale[J].Nature Methods,2014,11(3):333-337.
[16] Ashburner M,Ball C A,Blake J A,et al.Gene Ontology:tool for the unification of biology[J].Nature Genetics,2000,25(1):25-29.
[17] Jcquart P.Nouvelles recherches sur la distribution florale[J].Bull.Soc.Vand.Sci.Nat,1908(0):44.
[18] Finn R D.Pfam:the protein families database[J].Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics,2012.
[19] Apweiler R,Martin M J,O’Donovan C,et al.Update on activities at the Universal Protein Resource (UniProt) in 2013[J].Nucleic acids research,2013,41(D1):D43-D47.
[20] Smith T F,Waterman M S.Identification of common molecular subsequences[J].Journal of Molecular Biology,1981,147(1):195-197.
[21] Szklarczyk D,Franceschini A,Wyder S,et al.STRING v10:protein-protein interaction networks,integrated over the tree of life[J].Nucleic Acids Research,2014:gku1003.
[22] Yuan J,Wu W,Xie C,et al.NPInter v2. 0:an updated database of ncRNA interactions[J].Nucleic Acids Research,2014,42(D1):D104-D108.
[23] Xie C,Yuan J,Li H,et al.NONCODEv4:exploring the world of long non-coding RNA genes[J].Nucleic Acids Research,2014,42(D1):D98-D103.
RESEARCH ON PREDICTION ALGORITHM FOR LNCRNA-PROTEIN INTERACTIONSBASED ON HETEROGENEOUS NETWORK
Zheng Xiaoxiong Zhu Wenyan
(ShanghaiKeyLabofIntelligentInformationProcessing,SchoolofComputerScience,FudanUniversity,Shanghai200433,China)
Long noncoding RNA (lncRNA) plays a key role in biological and pathological processes. LncRNA can have interaction with multiple proteins, therefore predicting lncRNA-protein interaction comes to be one of the ways to study the functions of lncRNA. Because of the low conservation of lncRNA, the methods of predicting lncRNA-protein interaction by using machine learning algorithms with extracting the features of lncRNA and protein may be not fit enough. LPHeteSim algorithm is a method based on pair-wise random walk which can measure the relevance between lncRNA and protein in the lncRNA-protein heterogeneous network. In order to decrease the noise of protein similarity networks, we use similarity network fusion (SNF) algorithm to fuse the protein similarity networks under different metric together, and reach a better result.
LncRNA-protein interaction Randow walk Heterogeneous network
2016-03-14。郑肖雄,硕士,主研领域:生物信息。朱文琰,硕士。
TP3
A
10.3969/j.issn.1000-386x.2017.03.041
猜你喜欢
肝博士(2022年3期)2022-06-30
小学生学习指导(中年级)(2021年12期)2021-12-30
家庭教育报·教师论坛(2021年42期)2021-12-23
海外星云(2021年9期)2021-10-14
河北画报(2021年2期)2021-05-25
北方论丛(2021年2期)2021-05-22
汉字汉语研究(2020年2期)2020-08-13
电子制作(2019年22期)2020-01-14
疯狂英语·新读写(2018年3期)2018-11-29
公务员文萃(2013年5期)2013-03-11
