
我国开展药品上市许可持有人制度试点工作情况及相关政策建议
2017-04-07 06:20赵怀全北京积水潭医院药剂科北京100035
中国药房 2017年4期
赵怀全(北京积水潭医院药剂科,北京 100035)
·药业专论·
我国开展药品上市许可持有人制度试点工作情况及相关政策建议
赵怀全*(北京积水潭医院药剂科,北京 100035)
目的:了解并完善我国开展药品上市许可持有人制度试点工作。方法:分析我国药品上市许可制度存在的问题,以及美国、欧盟的药品上市许可制度的特点,归纳我国药品上市许可持有人制度试点的法律授权与内容,并对试点工作提出相关政策建议。结果与结论:我国药品“生产许可”与“上市许可”合并统一的药品上市许可制度,存在“新药”概念陈旧、药物资源浪费严重、新药研发创新动力不足等问题;而美国、欧盟施行的是药品“生产许可”与“上市许可”相分离的药品上市许可制度,其上市新药数量较多,产能利用率较高,药品安全、质量职责、风险管理等制度相对完善。我国有必要借鉴美国、欧盟的药品“生产许可”与“上市许可”相分离的管理制度,开展药品上市许可持有人制度试点工作。为了保障试点工作的有效开展,建议进一步加强和完善与药品安全管理相关的质量授权管理、药物警戒体系建立、药害救济制度建立等。
药品上市许可持有人制度;管理;试点工作
2015年8月18日,由国务院印发的《关于改革药品医疗器械审评审批制度的意见》[1](简称《意见》)中明确提出我国要开展药品上市许可持有人制度试点工作;随后于11月4日经全国人民代表大会常务委员会授权在北京、天津、河北、上海、江苏、浙江、福建、山东、广东、四川等10个省、市开展为期3年的试点[2]。长期以来,我国施行的是药品“生产许可”与“上市许可”合并统一的管理制度。按照2015年新修定的《中华人民共和国药品管理法》(简称《药品管理法》)及其配套法规的有关规定,药品批准文号只能颁发给拥有《药品生产许可证》的生产企业,取得药品批准文号后方可生产和上市销售药品。试行的药品上市许可持有人制度将药品的“生产许可”与“上市许可”相分离,鼓励药品创新,允许药品研发机构和科研人员取得药品批准文号,并对药品质量承担相应的责任。鉴于这种新的管理制度,全国人民代表大会常务委员会给予国务院相应的法律授权,国家食品药品监督管理总局(CFDA)为此也调整了相关政策,着力构建新的药品安全保障机制。为了充分认识这种新的管理制度,笔者结合我国药品上市许可制度存在的问题,比较我国药品上市许可制度和美国、欧盟药品上市许可制度的实现形式和特点,介绍我国药品上市许可持有人试点工作的法律授权与内容,并对试点工作提出相关管理建议。
1 我国药品上市许可制度存在的问题
依照《药品管理法》及其配套法规,我国药品上市许可制度施行的是药品“生产许可”与“上市许可”合并统一的管理制度,即只有持有《药品生产许可证》的企业才能申请注册生产和销售药品。随着我国医药产业的进一步发展升级,这一监管制度已不利于鼓励研发和创制新药,难以保护公民、法人和其他组织研发新药的合法权益,并出现了资源浪费、研发不足等问题,具体体现在以下方面:
1.1 “新药”概念陈旧
我国于2002年9月开始实施的《药品管理法实施条例》第83条指出,新药的含义是指未曾在中国境内上市销售的药品。这种以境域界定的“未曾在中国境内上市销售”的新药概念,导致即使在国外已经上市多年的药品在我国国内注册上市却变成了新药,即存在“新药不新”的状况。这是一种泛化的新药概念,扩大了新药的覆盖面,将本已不新的药品作为新药审批,不利于鼓励我国的新药研发。
1.2 仿制药水平较低
我国一直以仿制药为主,在2015年国家药品审评中心待审评的21 000个品种中,有90%是化学药仿制药[1]。自2007年10月1日起,我国施行的《药品注册管理办法》对仿制药作了如下定义:仿制药是指申请生产原国家食品药品监督管理局已批准上市的、已有国家标准的药品[3]。因部分药品质量标准水平不高,并缺乏药品质量和疗效一致性评价,使得一些仿制药的质量和美国、欧盟等相关产品的质量存在较大差距。
1.3 药品注册申报不规范
CFDA于2015年7月22日发布的《关于开展药物临床试验数据自查核查工作的公告》,要求企业对已申报生产或进口的1 622个待审药品注册申请开展药物临床试验数据核查工作。截至2015年12月31日,已有400多家企业主动撤回了951个药品注册申请;另外还有22家企业的24个品种被核查发现临床试验数据存在不真实、不完整的情况,不予批准注册。至此,被撤回、不予批准的批件数量占到了核查总数的60%[4]。
1.4 药物资源浪费严重
据CFDA公开的信息显示,我国现有国产药品批准文号(化学药、生物制品、中成药等)17万余个[5]。其中,含有同一成分不同剂型、包装、规格及其复方制剂的药品的重复现象十分严重。如含有阿司匹林(化学名为乙酰水杨酸)成分的药品批准文号有1 106个,含有对乙酰氨基酚成分的药品批准文号有3 071个,含有维生素C成分的药品批准文号有2 531个;仅板蓝根颗粒的批准文号就有1 035个,诺氟沙星胶囊的批准文号也多达721个。然而,目前市场上生产和使用的药品只涉及到5万多个批准文号,大量药品批准文号处于闲置状态,导致药物资源浪费。另外,三级综合医院基本用药供应目录规定:800张床位以上的医疗机构,所使用化学药品的品规数不能超出1 200个,中成药的品规数不能够超出300个[6]。可见,大量品规重复的药品批准文号已经缺乏市场价值,处于无产品生产的闲置状态,而很多企业还在继续申请注册。这种现象不仅浪费了大量审评、监管资源,也给药品招标、药品定价、医保管理、医疗机构药品遴选增加了巨大的成本投入。
1.5 新药研发创新动力不足
按照《药品注册管理办法》规定,药品研发机构或研发者只有通过采取技术转让的方式,将获得的研发成果如“新药证书”转让给生产企业才能实现药品的注册生产和销售。而药品上市所带来的收益多与研发者关联不大,特别是市场前景良好、技术含量高、后续开发潜力巨大的品种,无法实现相应的技术价值。由此可见,这种管理制度制约了药品研发机构或研发者的创新动力。另外,实施技术转让后,如药品的设计缺陷、质量提升、适应证调整、使用方法优化等与质量有关的问题,难以得到持续和系统的改进,相应的药品研发、生产、使用的安全责任也难以确认。
2 美国、欧盟的药品上市许可制度及其优势体现
目前,药品上市许可制度已成为国际上通行的药品准入管理制度[7]。美国和欧盟中的大多数成员国都是制药强国,其药品上市许可制度包括“上市许可”与“生产许可”两部分,特点是将药品“上市许可”与“生产许可”实行分离管理,有别于我国将两者授予同一主体,使得药品上市许可与生产企业不再捆绑,其经验值得借鉴。
2.1 美国、欧盟的药品上市许可制度
1938年6月,美国国会通过的《食品、药品和化妆品法案》第一次要求新药上市前必须提供安全证明;1962年,美国国会通过的《科夫沃-哈里斯修正案》建立了严格的药品上市前审批流程,并授权美国FDA加强对所有药品上市前的生产和销售监管,要求药品生产企业必须建立《药品生产质量管理规范》(GMP)后方可获得生产许可,接受药品上市许可持有人的委托生产;1984年,美国国会通过的《韦克斯曼-哈奇法案》是一个专门管理药品注册批准程序的特别法案,包括申请书的受理、新药技术评审、现场考察、通知审评结果、双方交流等。至今,美国的药品注册申请主要有2种[8]:一种是新药注册申请(New drug application),另一种是简化的药品注册申请(Abbreviated new drug application)(其申请依赖于已经通过审批的药品的试验数据,只需要进行生物等效性试验即可。大部分通用名药品的注册申请都属于此类,相当于我国的已经有国家标准的药品的申请)。美国这种长期形成和不断完善的药品“上市许可”和“生产许可”相分离的管理制度,促进了美国制药行业的快速发展。
欧盟的药品上市许可制度既考虑其整体性,又兼顾各成员国的自主性,经过多年发展已形成多部指令和条例相互配套、相互补充的法律体系。1965年,各成员国相互认可的第65号指令规定药品的上市程序为:(1)企业提交药品生产申请文件;(2)由主管部门进行评估,然后提供一个评估文件。随着监管的需要,欧盟委员会陆续颁布了一系列药事管理指令,如1991年的第356号指令制定了人用药品GMP;1993年的第2309号指令规定新药上市若是向欧盟申请的,发证后各成员国通用;2000年的第83号指令要求若在欧盟各成员国售药,必须符合欧盟原则。2001年,欧盟委员会对上述指令进行了梳理、整合,最终通过了第83号指令[9],作为欧盟全面、系统的药事法规。该法规对药品上市许可的概念和程序、标识和标签管理、药物警戒等作出了详细规定,如第6条规定:药品上市许可持有人应当为其上市的药品负责,承担药品上市后的安全信息、药物警戒、不良反应报告管理等。这些职责的规定,强化了药品上市许可持有人对药品上市的安全和质量管理的职能。2004年,欧盟委员会针对2001年83号指令中有关人用药品上市以及监管的部分内容进行修订,发布了726号指令,对药品上市许可的有效期和重新注册作出了规定;发布并实施了《欧盟人用药品风险管理制度指南》(2004年版),通过风险最小化评估、制订风险最小化计划,管控药品安全风险。欧盟的药品上市许可人和药品生产许可人可以是同一主体,也可以是两个独立的主体。只要药品注册申请人获批了某一产品的上市申请,便成为药品上市许可持有人,每种产品都有对应的药品上市许可持有人。
从美国和欧盟的药品上市许可制度来看,其法规条例、指令均较为详细、成熟、规范,明确规定了有关药品上市许可的程序,临床研究必须证明新药安全、有效,并确保质量可控,患者用药不会承担不合理的风险。
2.2 美国、欧盟药品上市许可制度的优势体现
新药研发的上市数量和产能利用率(也称设备利用率,就是指实际生产能力到底有多少在运转及发挥作用),可在一定程度上体现药品上市许可制度对制药行业发展的影响[10]。新药研发数量会受到药品上市许可制度的制约,当两者之间相适应时,新药的上市数量就相对较多,同时产能利用率也越高,实际生产能力运转及发挥的作用越好;反之,新药研发上市数量就相对较少,产能利用率也相对较低。据相关研究显示,2009-2013年我国医药制造业的产能利用率均值仅为62%,相比美国、欧盟80%以上的产能利用率,我国医药制造业产能严重过剩,其原因与药品上市许可制度的差异不无关系。我国创新药上市数目过少,平均每年不足10个,远远落后于欧美发达国家平均每年的20~100个[10]。2009-2013年我国与美国、欧盟医药制造业产能利用率比较见图1;2007-2014年我国与美国、欧盟上市新药数量比较见图2。
分析图1、图2可知,美国、欧盟通过实施药品“上市许可”和“生产许可”相分离的管理制度,其新药研发上市数量和产能利用率均明显要高于我国,显示出了一定的制度优势。同时也表明,药品上市许可持有人与是否拥有生产许可没有必然联系,申请人可以只具备投资研发或自行研发的能力,也可以与具备生产许可的企业合作,这样就可有效提升新药的研发能力和产能利用率。

图1 2009-2013年我国与美国、欧盟医药制造业产能利用率比较Fig 1 Comparison of utilization rate of pharmaceuticalmanufacturingcapacityamongChina,USAand EU during 2009-2013
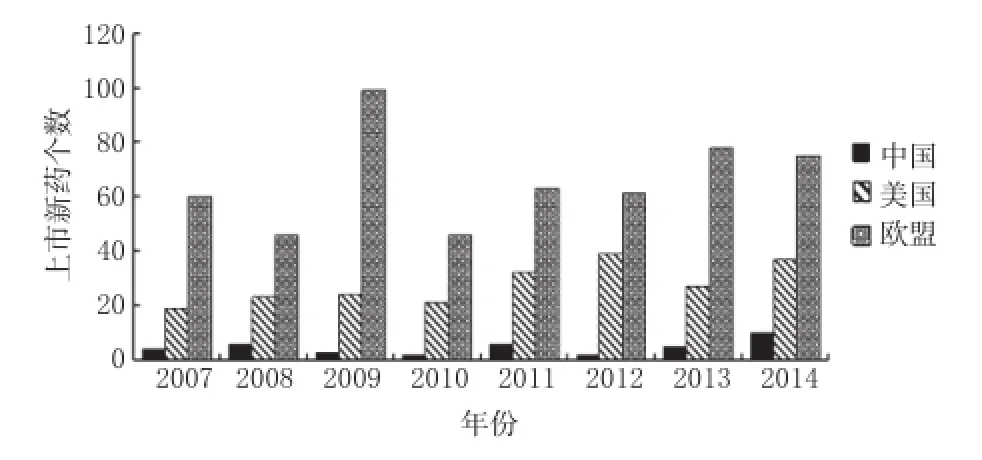
图2 2007-2014年我国与美国、欧盟上市新药数量比较Fig 2 Comparison of the number of new marketed medicines among China,USA and EU during 2007-2014
3 我国开展药品上市许可持有人制度试点工作情况
《意见》首次提出我国开始实施药品上市许可持有人制度试点工作,推进药品审批制度改革。同时,重新修订了新药的概念,将新药的概念由现行的“未曾在中国境内上市销售的药品”调整为“未在中国境内外上市销售的药品”,鼓励新药研发和创新,提高药品质量,为建立新的药品准入管理制度提供了政策支持。
3.1 全国人大常委会授权国务院开展药品上市许可持有人制度试点工作
2015年11月4日,全国人民代表大会常务委员会决定授权国务院在北京、天津、河北、上海等10个省、市开展药品上市许可持有人制度试点工作,试行药品“上市许可”与“生产许可”相分离的管理制度。时间自2015年11月5日起至2018年11月4日止,实施期限为3年。如果试点工作的内容经实践证明是可行的,将修改和完善《药品管理法》有关药品上市许可管理的相关条款。这项授权在法律层面为我国开展药品上市许可持有人制度试点工作提供了充分的法律保障,体现了依法治国和依法行政的基本原则。
3.2 推进药品上市许可持有人制度政策实施
为了积极推进我国药品上市许可持有人制度试点工作,2015年12月17日国务院批复CFDA,建立药品、医疗器械审评审批制度改革部际联席会议制度[11],加强协调指导,及时制定推进改革的政策措施。随即,北京市于2016年1月率先启动药品上市许可持有人制度试点工作,并得到了CFDA给予的审评审批、绿色通道等12项政策支持[12]。上海也已选定首个生物制药合同生产(CMO)试点企业[13],开展药品上市许可持有人制度;该企业不仅可提供先进的生物制药技术、委托生产管理体系、质量保障体系以及产品的供应链管理服务,还可与药物研发企业和机构合作,促进中国创新药物的临床研究与上市,推动中国企业生物技术药物进入国际市场。如上所述,药品上市许可持有人制度试点改革的多项政策,正在我国逐步开始启动和实施。
3.3 药品研发机构和科研人员可取得药品批准文号
为了鼓励科研人员、研发单位和企业创新研发新药,《意见》将申请注册药品的范围进行了调整和扩大,按照药品“上市许可”与“生产许可”相分离的管理制度,规定科研人员、研发单位研发成功后也可发给批准文号,施行药品批准文号持有人和生产企业相分离的制度,让更多的科研人员投入到创新队伍和行业当中来,吸引更多的资本投入,推动成果转化和医药产业升级。同时,减少一些重复建设,优化产业结构调整,提升医药产业的综合竞争力。
3.4 药品上市许可持有人对药品质量承担相应的责任
2016年5月26日,国务院办公厅印发的《药品上市许可持有人制度试点方案》明确了药品注册申请人和药品上市许可持有人应承担的法律责任和义务,药品注册申请人和药品上市许可人应对药品申请中提交的所有资料的真实性和准确性负责[14]。药品注册申请人获得药品的上市许可后,可委托不同的生产企业生产药品,药品的安全性、有效性和质量可控性由上市许可人负责。无论其是否将药品委托给其他企业生产,发生药品不良事件后,药品上市许可持有人都要承担相应的民事或刑事责任,然后依据合同约定对生产企业进行追责。药品上市许可持有人应定期分析、评估药品的安全性信息,撰写“定期安全性更新报告”,发布与药品安全相关的警戒信息,保障药品质量和患者用药安全。
4 完善我国药品上市许可持有人制度试点工作的相关政策建议
我国开展药品上市许可持有人制度试点是一项创新性工作,有关药品法律责任追究、药品监管制度、药品技术转让体系和药品委托生产等方面的政策均面临着调整,相应的管理措施也亟待加强和完善,以保障药品上市许可持有人制度试点工作的正常开展。笔者认为,为了保障试点工作的有效开展,尽管可借鉴美国、欧盟的药品上市许可持有人制度的相关经验,但还必须考虑到我国的法律环境和药品监管实际,构建和完善相应的管理体系,保证药品质量,保障公众的健康权益。对此,特提出如下建议:
4.1 完善药品质量授权人管理制度
实施药品上市许可持有人制度之后,许可持有人就成了药品整个生命周期的质量责任主体,需要负责对药品的研发、审批、生产、上市的全过程进行质量监控。特别是在药品生产环节,为了确保上市药品的质量安全,许可持有人需要全面负责物料、生产工艺、质量标准的审核,甚至每个批次产品的放行等。药品质量授权人管理制度在药品安全管理中具有重要的作用,《欧盟人用药品风险管理制度指南》(2004年版)曾针对药品上市许可人提出了一系列指导性建议,如怎样规范地开展药品整个生命周期的风险管理、风险最小化评估与计划以及怎样保障药品的质量与安全等。因此,我国有必要借鉴欧盟的做法,在开展药品上市许可持有人制度试点工作中,进一步完善与之配套的药品质量授权人管理制度,明确管理内涵,并将其纳入药事管理相关法规中,赋予该项制度相应的法律保障,使之得到有效实施。
4.2 完善药物警戒体系建设
长期以来,我国药品监督管理部门和药品不良反应监测机构承担着药品的风险信息收集、评价、干预等职责。国家药品不良反应监测中心的监测数据是风险信号的主要来源,根据这些信号,主管部门分别有针对性地采取某些干预措施,如责令停止药品生产、销售和使用,责令修改药品说明书、标签等干预措施。随着药品上市许可持有人制度试点工作的开展,药品上市许可持有人将呈现数量扩大和结构多样化的变化,提高药物警戒管理能力成为必然。因此,亟待建立药品上市许可持有人、药品生产和经营企业参与的药物风险管理制度,发现、评价、认识和预防药品不良反应及其他药物相关问题。由于我国目前的药事法规尚未对“药物警戒”进行明确定义,为实施药品上市许可持有人制度,监测药品整个生命周期的风险信号,需要相应完善我国的药物警戒体系建设,从制度和机制层面完善药品安全管理体系。
4.3 探索并建立药害救济制度
实施药品上市许可持有人制度,上市许可持有人须相应承担药品安全性、有效性和质量保障的法定责任。现实中,药品质量缺陷、药品不良反应是客观存在的。近几年来,我国相继发生过“齐二药”假药事件、“欣弗”质量事件、“龙胆泻肝丸”肾损害事件等,损害了公众的生命安全和健康权益,造成了严重的社会影响。
由药品原因导致的伤害包括药品质量缺陷导致他人的人身损害、药品不良反应导致他人的人身损害等。当中小企业、科研人员、研发机构成为药品上市许可持有人时,必须建立相应的保障机制和救济制度,使其有能力承担伤害救助职责,使由于药品质量缺陷导致损伤的受害人真正获得救济;同时,对受害人用药后的不良反应损害提供救济。我国目前的法规中,均缺乏对药害救济制度的相关规定。因此,明确药品上市许可持有人产品责任的同时,需要建立药害救济赔偿制度。
5 结语
我国开展药品上市许可持有人制度试点是一项全新的工作,必将对药品研发、产业发展产生重要而深远的影响。该试点工作授权期限仅有3年,相对于药物研发周期、注册申报时限、配套制度的建立等来说时间是紧迫的。鉴于美国、欧盟的药品上市许可制度的经验,试点省、市有必要围绕药品“上市许可”与“生产许可”分离管理的核心内涵,推动管理制度创新,把握重要的发展机遇。同时,在试点工作中加强配套制度建设,实施药品整个生命周期的安全管理,始终将“确保药品质量、保障公众生命健康权益”放在首位,并及时对试点工作的效果进行评估,将药品上市许可持有人制度试点成果向制度成果转化,完善我国的药品监管制度,提高新药研发和药品生产的质量和效益。
[1] 国家食品药品监督管理总局.国务院关于改革药品医疗器械审评审批制度的意见[EB/OL].(2015-08-18)[2016-05-03].http://www.sda.gov.cn/WS01/CL0056/126821.html.
[2] 全国人民代表大会常务委员会.关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定[S].(2015-11-04)[2016-05-03].http://www.sda.gov.cn/WS01/CL0051/133921.html.
[3] 国家食品药品监督管理局.药品注册管理办法[S].2007-10-01.
[4] 国家食品药品监督管理总局.关于154家企业撤回224个药品注册申请的公告[EB/OL].(2015-12-31)[2016-05-03].http://www.sfda.gov.cn/WS01/CL0087/140440.html.
[5] 国家食品药品监督管理总局.数据查询[EB/OL].(2016-02-10)[2016-05-03].http://app1.sfda.gov.cn/datasearch/face3/dir.html.
[6] 国家卫生和计划生育委员会.三级综合医院评审标准实施细则:2011年版[S].2011-12-23.
[7] 邵蓉,郑澜,胡晨希,等.我国建立药品上市许可人制度的必要性与可行性分析:基于制度变迁均衡分析模型[J].中国药房,2014,25(33):3076-3080.
[8] 韦冠.国内外药品上市许可制度比较及借鉴[J].中国药房,2008,19(34):2650-2653.
[9] 张旭.药品安全:生产商不能承受之重:从欧盟看药品上市许可管理制度的起源和发展[J].中国食品药品监管,2012(7):51-54.
[10] 陈永法,伍琳,邵蓉.药品上市许可与生产许可分离的新制度经济理论研究[J].中国医药工业杂志,2015,46(9):1034-1039.
[11] 国务院.关于同意建立药品医疗器械审评审批制度改革部际联席会议制度的批复[EB/OL].(2015-12-24)[2016-05-03].http://www.gov.cn/zhengce/xxgkzl.htm.
[12] 马昊楠,许方霄.北京市率先启动药品上市许可持有人制度试点工作:中关村医药产业发展获国家12条政策支持[J].首都食品与医药,2016(3):8.
[13] 国内首个生物制药CMO试点项目“花落”张江[EB/OL].(2016-02-17)[2016-05-03].http://sh.people.com.cn/n2/2016/0217/c134768-27748625.html.
[14] 国务院办公厅.关于印发药品上市许可持有人制度试点方案的通知[S].2016-05-26.
(编辑:杨小军)
Recommendation on Pilot Work and Related Policies of Medicine Marketing Authorization Holder System in China
ZHAO Huaiquan(Dept.of Pharmacy,Beijing Jishuitan Hospital,Beijing 100035,China)
OBJECTIVE:To investigate and perfect the pilot work of medicine marketing authorization holder system in China. METHODS:The problems of medicine marketing authorization system in China and the features of medicine marketing licensing system in USA and EU were analyzed.The legal authority and content of pilot medicine marketing authorization holder system in China were summarized so as to propose related policy recommendation.RESULTS&CONCLUSIONS:There are many problems in the medicine authorization system in China which is the combination of“production license”and“marketing license”,for example,the obsolete definition of“new drugs”,the serious waste of drug resources,the insufficient research and development of new drugs.The“production license”and“marketing license”are separated in the USA and EU.There are many new drugs,high capacity utilization and perfect system of drug safety,quality control and risk management.In order to ensure the effective implementation of the pilot work,it is suggested to enhance and improve the quality authorization management,the pharmacovigilance system and drug injury relief related to drug safety management.
Medicine marketing authorization holder system;Management;Pilot work
R95
A
1001-0408(2017)04-0433-05
2016-05-03
2016-08-30)
*主任药师。研究方向:医院药学、药事管理。电话:010-58516679。E-mail:jsthuaiquan@163.com
DOI10.6039/j.issn.1001-0408.2017.04.01
猜你喜欢
华人时刊(2022年1期)2022-04-26
中国食品药品监管(2022年1期)2022-03-03
今日农业(2021年12期)2021-11-28
支部建设(2019年36期)2019-02-20
少先队活动(2018年10期)2018-12-29
人大建设(2017年4期)2017-07-21
妇女生活(2015年4期)2015-09-10
创业家(2015年9期)2015-02-27
创业家(2015年9期)2015-02-27
创业家(2015年9期)2015-02-27
