
几种天然产物与CASP3靶点的相互作用机制探索*
2017-03-27 09:18:13张静晓刘晓洁陈盼盼张丽雷
世界科学技术-中医药现代化 2017年11期
张静晓,刘晓洁,杨 春,陈盼盼,张丽雷
(湖北民族学院化学与环境工程学院 恩施 445000)
细胞凋亡存在于多细胞生物的整个生命过程中,此过程可及时清除机体内多余和受损伤的细胞,维持组织器官的稳定性,对机体自身的稳定具有关键的作用[1]。细胞的凋亡受到生物体自身代谢的严格调控,当细胞的正常凋亡过程受到干扰,细胞便会产生异常,从而出现特殊表型,细胞分化停滞甚至产生肿瘤[2]。参与细胞凋亡过程的主要包括凋亡蛋白酶(caspases)、衔接蛋白(adapter proteins)、Bcl-2和凋亡抑制蛋白(IAPs)等4类蛋白。其中Caspase半胱氨酸蛋白酶家族包含高度同源的半胱氨酸蛋白酶,分为两个主要的亚科,一个亚科家族参与炎症过程,包括CASP1,4和5等,另一个亚科家族在细胞凋亡过程中起重要作用,包含CASP3,8和9等[3]。CASP3的作用方式是,当细胞受到损伤时,其主要表现为线粒体膜损伤,线粒体内的细胞色素释放到细胞外,激活CASP3从而诱导细胞凋亡[4]。CASP3作为治疗某些癌症的靶点受到越来越多的关注,研究CASP3抑制剂是抗癌药物开发的有效方法之一。
基于生物信息学方法,从天然产物中筛选有效成分,研究其与靶点的作用机制,是新药开发和研究中药作用机理的重要方法[5-8]。本课题组利用多种生物信息学方法建立了一个中药有效成分筛选的模拟体系,通过类药性筛选、靶点预测等方法,从中草药中筛选出与CASP3靶点有相互作用的7种有效成分[9],如表1所示。本文在前期研究的基础上,采用Autodock Vina程序[10]对筛选后的有效成分与CASP3进行分子对接,之后进行分子动力学(MD)模拟,通过分析分子模拟的轨迹以及得到的相互作用能,预测它们与CASP3的结合位点以及作用方式。
1 材料和方法
1.1 材料的准备
从蛋白晶体数据库(RCSB PDB数据库)中得到CASP3靶点的晶体结构(PDB ID:5I9T),除去晶体水,并对其加氢,作为分子模拟时受体的初始模型,记为CASP3。天然产物中的药物分子从化学结构数据库(Chemspider)中获取,采用GAMESS量子化学软件包[11],在B3LYP方法,6-31(d,p)基组水平上优化得到药物分子的结构,通过频率分析保证其处于稳定结构,作为分子对接时配体的初始构象。
1.2 分子对接
药物分子与CASP3的对接在AutoDock Vina程序中进行,使用半柔性对接方法,CASP3被视为一个刚体和药物分子所有的可旋转键进行对接。最有可能的结合位点采用搜索全局优化法(Iterated Local Search Globle Optimizer)搜索。最佳的结合位点设定为涵盖CASP3整个活性口袋大小为30×30×30 Å3的盒子内,盒子的网格间距为1.0 Å,以药物分子的几何中心为中心,在每个对接过程中,根据在AutoDock Vina的评分函数计算出的结合亲和能,筛选出了10个打分最高的构象模型。
1.3 分子动力学模拟
分子动力学(MD)模拟在NAMD(版本2.11)程序[12]中进行,将分子对接得到的复合物结构用作MD模拟的初始构象,采用CHARMM 27全原子力场。药物配体分子力场参数采用SwissParam程序生成。在复合物的周围建立可将复合体完全包围且延伸8 Å的立方体水模型的周期性结构,原子的总数目大约为12 400个。在进行分子动力学模拟之前,进行两步优化,使整个体系松弛:第一步,用最陡下降法和共轭梯度法优化水分子;第二步,将整个体系优化至收敛。优化结束后,为了避免快速升温对溶质分子带来的影响,采取了缓慢升温的方法,体系温度在220 ps内由0 K缓慢加热至310 K,之后进行10 ns的NPT模拟,时间步长为2 fs。在动力学模拟过程中,应用Langevin动力学控制温度,碰撞频率为1.0 ps-1。将所有和氢原子相连的键设置为不振动,使用PME(Particle Mesh Ewald)方法计算长程静电相互作用,范德华相互作用的截断值为12 Å。
2 结果与讨论
2.1 分子对接
将本课题组筛选得到的7个与CASP3靶点有相互作用的化合物为研究对象[7],分别与CASP3进行对接,通过AutoDock Vina程序对对接结果进行打分,所得分值如表1所示。由表可见,丹参酮IIA和野黄芩苷与CASP3具有较好的空间匹配度,所需结合能量最低。
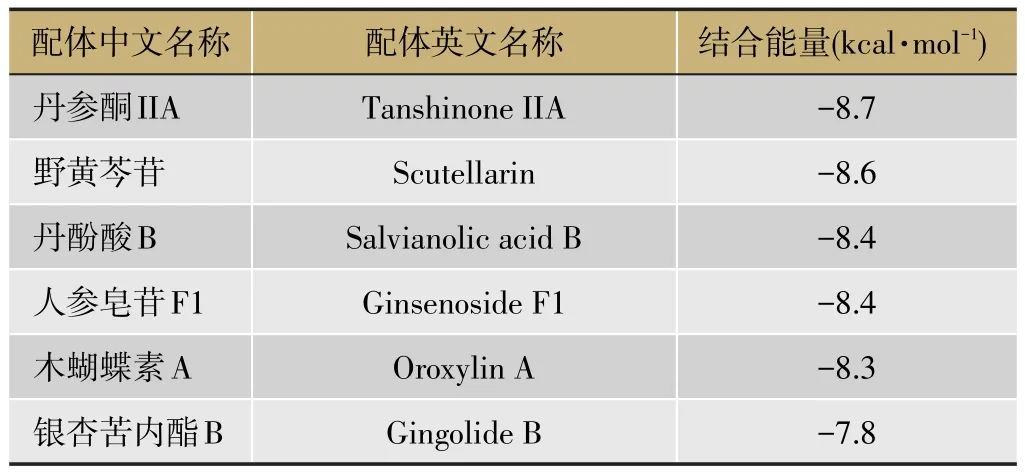
表1 分子对接的最优化模型结果
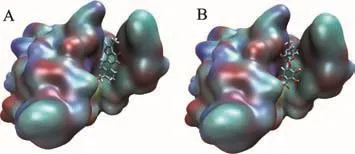
图1 丹参酮IIA(A)、野黄芩苷(B)与CASP3模型对接后的结果
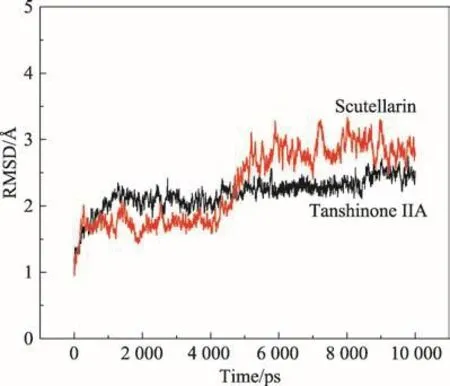
图2 CASP3复合物模型MD模拟期间骨架原子的均方根偏差
丹参酮IIA(A)、野黄芩苷(B)与CASP3对接结果如图1所示。从图中可以看出丹参酮IIA、野黄芩苷均对接在CASP3的活性口袋中,参考X射线衍射法获得的5I9T模型,对接的位置是合理的。
2.2 分子动力学
将丹参酮IIA、野黄芩苷的对接结构作为初始构象,进行了10 ns的分子动力学模拟,对其进行RMSD计算,结果如图2所示。从图中可见,这两个体系骨架原子的RMSD逐渐趋于稳定,其中丹参酮IIA与CASP3结合的构象很快达到稳定,最后上下波动幅度稳定在0.5 Å左右,野黄芩苷与CASP3结合的构象在5 ns左右经历一个上升后达到稳定,上下波动幅度稳定在0.7 Å左右。
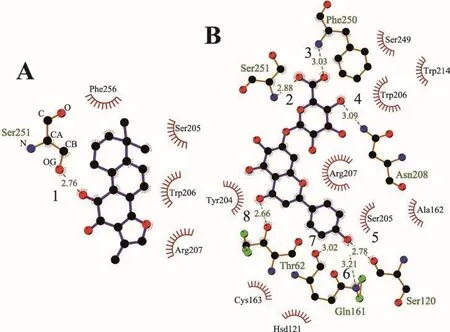
图3 丹参酮IIA(A)和野黄芩苷(B)与CASP3模型的相互作用网络
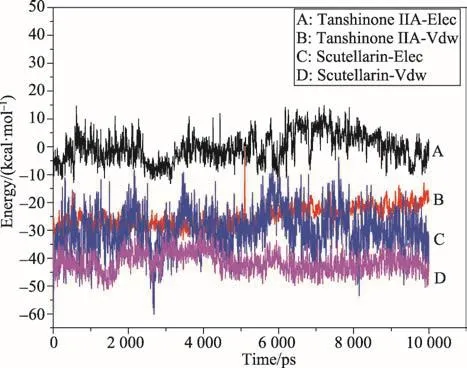
图4 丹参酮IIA(A)和野黄芩苷(B)与CASP3的相互作用力(VDW:范德华力,Elec:静电力)
在最后的1 000 ps的运动轨迹中,取最低能量结构进行了相互作用分析。使用软件Ligplot+[13]计算得到丹参酮IIA和野黄芩苷与CASP3模型的相互作用网络,如图3所示。从图中可见,丹参酮IIA和野黄芩苷均与CASP3形成疏水相互作用,其中丹参酮IIA与4个残基(Phe256、Ser205、Trp206、Arg207)具有疏水作用,并且与Ser251形成一个键长为2.76 Å的氢键;野黄芩苷与9个残基(Ser249、Trp214、Trp206、Arg207、Ser205、Ala162、Tyr204、Cys163、Hsd121)有 疏 水 作 用 ,与Ser251和Phe250等6个残基形成7个氢键。其中,与残基Ser251的O原子形成的氢键键长为2.88 Å,与残基Phe250的N原子形成的氢键键长为3.03 Å,与残基Asn208的N原子形成的氢键键长为3.09 Å,与残基Ser120的O原子形成的氢键键长为2.78 Å,与残基Gln161的N原子和O原子分别形成2个氢键,键长分别为3.21 Å和3.02 Å,与残基Thr62的O原子形成的氢键键长为2.66 Å。由于蛋白质和药物分子之间形成的氢键有利于它们的结合,说明野黄芩苷与CASP3的结合能力优于丹参酮IIA与蛋白质的结合能力。
对丹参酮IIA和野黄芩苷分别与CASP3形成的静电力和范德华力进行了分析,结果如图4所示。从图中可见,配体分子与CASP3之间的作用力在不同模拟时间较为稳定。丹参酮IIA与CASP3之间的范德华力平均值为-21.97 kcal·mol-1,静电力平均值为-5.31 kcal·mol-1,故主要作用力为范德华力;野黄芩苷与CASP3之间的范德华力平均值为-41.39 kcal·mol-1,静电力平均值-26.51 kcal·mol-1,故其主要作用力也为范德华力。野黄芩苷与CASP3之间的结合强度大于丹参酮IIA,这是由于它与CASP3之间不仅具有较大的范德华力,而且具有较强的静电力,与前述野黄芩苷与CASP3形成了较多的氢键的结论一致。
2.3 氢键分析
氢键是维系蛋白质与配体分子稳定性的重要作用力[14],分别对丹参酮IIA和野黄芩苷与CASP3形成的几条氢键进行分析,判断氢键采用的几何依据为氢键键长不大于3.5 Å,键角不大于30°,以5 000 ps到10 000 ps的运动轨迹为研究对象,计算了平均键长,平均键角,以及键长存活概率(氢键长度小于3.5 Å,同时键角小于30°的概率),结果如表2所示,表中氢键编号如图3所示。
由表2和图3可见,丹参酮IIA与CASP3形成的一个氢键不太稳定,存活率为48.03%。野黄芩苷与CASP3形成的七个氢键中,与残基Ser251的O原子形成的氢键(编号2),与残基Asn208的N原子形成的氢键(编号4),与残基Gln161的N原子形成的氢键(编号6),与残基Thr62的O原子形成的氢键(编号8)均不稳定,存活率分别为2.35%、10.04%、3.10%和7.35%。然而,与残基Phe250的N原子形成的氢键(编号3),与残基Ser120的O原子形成的氢键(编号5),与残基Gln161的O原子形成的氢键(编号7)的存活率分别为90.15%、85.16%和82.31%,表明这些氢键较为稳定,是野黄芩苷与CASP3形成较强相互作用的主要原因。
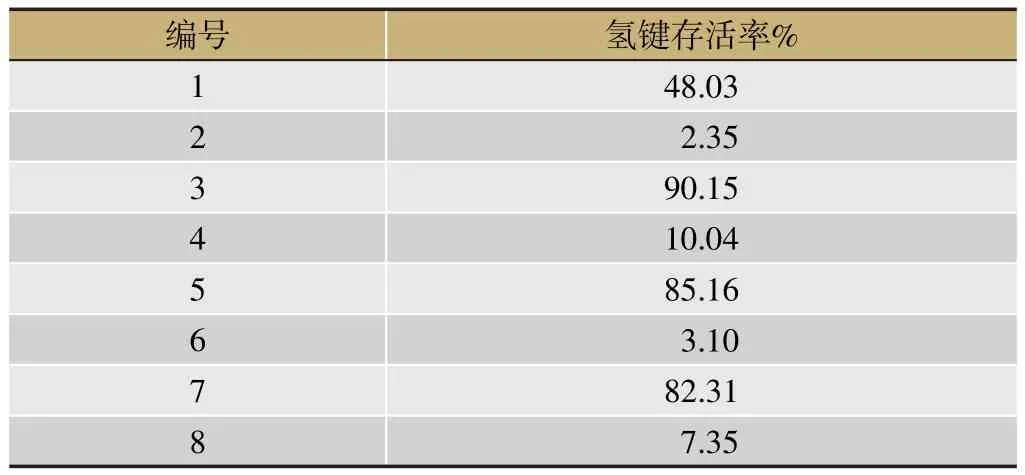
表2 氢键情况统计表
3 结论
本文以从几种天然产物的化学成分中筛选出的小分子为研究对象,以分子对接和分子动力学方法,研究了其与CASP3之间的相互作用。结果发现,丹参酮IIA和野黄芩苷具有较好的对接结果,并通过分子动力学方法获取了这两种药物与CASP3结合的稳定结构,它们之间的范德华力均强于静电力。另外,丹参酮IIA与CASP3中的Phe256、Ser205、Trp206等4个氨基酸残基具有疏水作用,形成1个氢键。野黄芩苷与CASP3靶点中的Ser249、Trp214、Trp206等9个氨基酸残基具有疏水作用,形成了7个稳定性不同的氢键,其中与残基Phe250的N原子,与残基Ser120的O原子,与残基Gln161的O原子形成的3个氢键最为稳定,是形成稳定构象的主要原因。研究与其相似的结构,有望获得CASP3靶点的有效抑制剂。目前,相关研究正在继续展开。
1 李敏,林俊.细胞凋亡途径及其机制.国际妇产科学杂志,2014,41(2):103-107.
2 Zeng C,Xing R,Liu J,et al.Role of CSL-dependent and independent Notch signaling pathways in cell apoptosis.Apoptosis,2016,21(1):1-12.
3 Lin B,Zhu M,Wang W,et al.Structural basis for alpha fetoproteinmediated inhibition of caspase-3 activity in hepatocellular carcinoma cells.Int J Cancer,2017,141(7):1413-1421.
4 Lakhani S A,Masud A,Kuida K,et al.Caspases 3 and 7:key mediators of mitochondrial events of apoptosis.Science,2006,311(5762):847-851.
5 叶小彤.基于生物信息学的中药蛋白质成分作用机制研究.北京:北京中医药大学硕士学位论文,2017.
6 周玥.蛋白激酶CK2抑制剂作用机理的分子模拟研究及其靶向设计.北京:北京工业大学博士学位论文,2016.
7 Musyoka T M,Kanzi A M,Lobb K A,et al.Structure based docking and molecular dynamic studies of plasmodial cysteine proteases against a South African natural compound and its analogs.Scientific reports,2016,6:23690.
8 Aiya Subramani P,Mahendran R,Dinakaran Michael R.Molecular Docking and Dynamics Simulation of Vibrio anguillarum Aspartate Semialdehyde Dehydrogenase with Natural Product Caulerpin.Lett Drug Des Discov,2016,13(3):255-261.
9 Zhang J,Li Y,Chen X,et al.Systems pharmacology dissection of multiscale mechanisms of action for herbal medicines in stroke treatment and prevention.Plos One,2014,9(8):e102506.
10 Trott O,Olson A J.AutoDock Vina:Improving the speed and accuracy of docking with a new scoring function,efficient optimization,and multithreading.J Comput Chem,2010,31(2):455-461.
11 Schmidt M W,Baldridge K K,Boatz J A,et al.General atomic and molecular electronic structure system.J Comput Chem,1993,14(11):1347-1363.
12 James C P,Rosemary B,Wei W,et al.Scalable molecular dynamics with NAMD.J Comput Chem,2005,26(16):1781-1802.
13 Laskowski R A,Swindells M B.LigPlot+:multiple ligand-protein interaction diagrams for drug discovery.J Chem Inf Model,2011,51(10):2778-2786.
14 Nick P C,Scholtz J M,Grimsley G R.Forces stabilizing proteins.FEBS letters,2014,588(14):2177-2184.
猜你喜欢
吉林大学学报(理学版)(2025年1期)2025-02-06 00:00:00
电子产品可靠性与环境试验(2023年5期)2023-06-04 16:35:39
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
科学技术创新(2022年36期)2022-02-13 04:29:09
科学技术创新(2022年36期)2022-02-01 10:22:22
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
中国机械工程(2019年7期)2019-04-23 07:14:32
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
池州学院学报(2015年3期)2016-01-05 01:13:04
中学化学(2015年8期)2015-12-29 07:32:44
