
ZnSe和CuXSe2(X=Al,Ga,In)结构和光学性质的研究
2017-03-27 07:20董玉静高延利
重庆工商大学学报(自然科学版) 2017年2期
董玉静, 高延利
(信阳学院 理工学院,河南 信阳 464000)
ZnSe和CuXSe2(X=Al,Ga,In)结构和光学性质的研究
董玉静, 高延利
(信阳学院 理工学院,河南 信阳 464000)
利用赝势平面波基组的密度泛函理论方法,首先优化太阳能材料ZnSe和CuXSe2(X=Al,Ga,In)的晶体结构,得到晶格参数、键长,并预测了CuXSe2带隙和光学性质,带隙按照Al→Ga→In依次减小,但晶格参数和形变参数依次增加;通过光学性质中介电函数、吸收系数,反射率和光电导率分析发现,吸收系数的最强峰都在紫外区域,在3种晶体中光学性能按照Al→Ga→In依次增强。
密度泛函方法;半导体;带隙;光学性质
在各种薄膜光伏电池材料中,铜基材料具有许多独特的优势,在光伏电池领域有着巨大应用潜力,从20世纪70年代就开始出现了大量的理论研究工作。近年来,由于光伏发电技术日趋得到重视,世界范围内大量课题组[1-9]相继开始了相关研究。
在20世纪70、80年代后的很长一段时间,人们将注意力转向铜基材料缺陷性质和能带匹配等侧重实际应用的研究方向。随着理论方法的改进,近几年又出现大量在铜基多元半导体的基本电子结构性质相关方面的研究报道。2009年Chen S等人[10]研究了II-VI二元半导体到 I-III-VI2三元半导体再到I2-II-IV-VI4或者I-III-II2-VI4四元半导体(I = Cu、Ag;II = Zn、Cd;III = Al、Ga、In;IV=Ge、Sn;VI = S、Se、Te)的性质演化规律。
目前已有大量采用密度泛函方法对铜基多元半导体的相关研究,但是关于这类材料的基本电子结构性质以及电子在结构性质影响的理论结果和实验结果存在大量争议。需要从半导体材料的根本出发,研究电子在二元半导体到三元半导体演化过程中的影响和作用,建立晶体结构并提供可靠的计算参数。先对ZnSe和CuXSe2(X=Al,Ga,In)的结构优化,考察电子在晶体结构的影响,然后通过电子在能带结构和态密度的影响进行分析,对二元半导体和三元半导体进行对比,最后重点分析三元半导体的光学性质。
1 ZnSe 和CuXSe2(X=Al,In,Ga)半导体的晶体结构
II-VI二元、Cu-III-VI2三元晶体结构和电子结构存在众多相似性,首先以ZnSe为例研究二元半导体中优化晶体结构,分析电子在结构和带隙中的影响,进一步研究分析铜基材料电子的性质及其对晶体结构、电子结构以及光学性质的影响。


图1 ZnSe的晶体结构图Fig.1 Crystal structure of ZnSe
2 计算模型和方法
在计算过程中采用广义梯度近似的GGA-PBE(Perdew-Burke-Ernerhof)[11]泛函方法处理交换-关联能,通过一组平面波基矢对电子波函数进行展开,采用超软赝势来描述离子实与价电子的相互作用。为满足计算精度并尽量确保计算速度,在k空间中,平面波截止能量(cut-offenergy)取650eV,采用4×4×4的k点网格[12-13]对全布里渊区求和。二元半导体的ZnSe价态电子包括Zn原子的3d104s2电子和Se原子的4s24p4电子。三元半导体中Cu原子为3d104s1电子,Se原子为4s24p4电子,Al原子为3s23p1电子,Ga原子为3d104s24p1电子和In原子为4d105s25p1电子。
3 结果与讨论
3.1 结构优化和带隙值
首先对二元半导体ZnSe和三元半导体CuXSe2(X=Al,Ga,In)结构进行优化,并对晶格参数计算值和实验值进行对比(表1),发现在计算的结果中,晶格参数和实验值误差都很小,说明计算方法是可行的,计算结果是可靠的。ZnSe因为有高度的对称性,在计算结果中形变参数是0.5。而在三元半导体CuXSe2(X=Al,Ga,In)中,随着原子序数的增加,形变参数增加,η<1 表示晶体在(001)方向被压缩。由结果可知原子序数增加,压缩程度减小。
在两类晶体电子结构中带隙的结果可以看出来,计算值比实验值明显偏小,这一般认为导致这种偏差的原因主要是采用广义梯度近似(或局域密度近似)下的密度泛函理论计算基态能带时,对电子与电子之间的交换—关联能作用处理不足引起的[14]结果不影响对晶体其他性质的研究和分析。
在对ZnSe和三元铜基CuXSe2(X=Al,Ga,In)半导体材料结构优化的基础上,计算其能带带隙,此两类材料都属于直接带隙半导体材料。在表1中可以发现ZnSe的理论值和实验值符合很好,而三元半导体材料中计算值比实验值小得多,且CuXSe2(X=Al,Ga,In)的带隙值按照Al→Ga→In的顺序减小,和结构参数的变化相反。
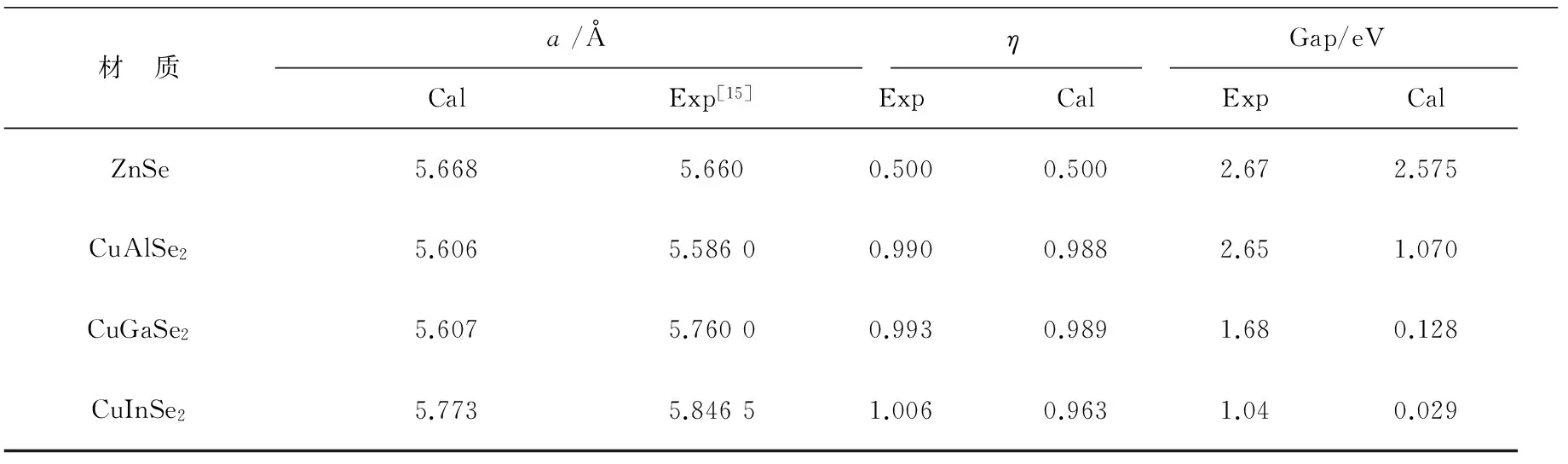
表1 ZnSe和CuXSe2(X=Al,Ga,In)晶胞参数以及带隙的实验值与计算值
3.2 光学性质


图3 CuXSe2(X=Al,Ga,In)晶体复介电函数实部和虚部变化曲线Fig3.Variation curves of the real and imaginary parts of dielectric function of CuXSe2(X=Al,Ga,In)crystals
复介电函数的虚部是连接材料晶体结构内部带间跃迁的微观物理过程和固体内部电子结构的桥梁。图3是CuXSe2体系介电函数实部和虚部的变化曲线。通过分析实部变化曲线发现在较低能量区域内, X=Al,Ga介电函数实部逐渐增大达到峰值,分别为7.96和10.55,这和CuAlX2(X=S, Se, Te)[17]的变化趋势比较近似,但CuInSe2却出现减小的趋势,这和冯晶[18]研究的结果是一样的。而实部变化却逐渐减小,出现最小值。此最小值按照Al→Ga→In的顺序朝低能量方向移动,依次分别为9.48、9.27和7.44。此外,在零频时3种材料的平均静态介电常数随着X原子从Al到In的顺序逐渐增大。
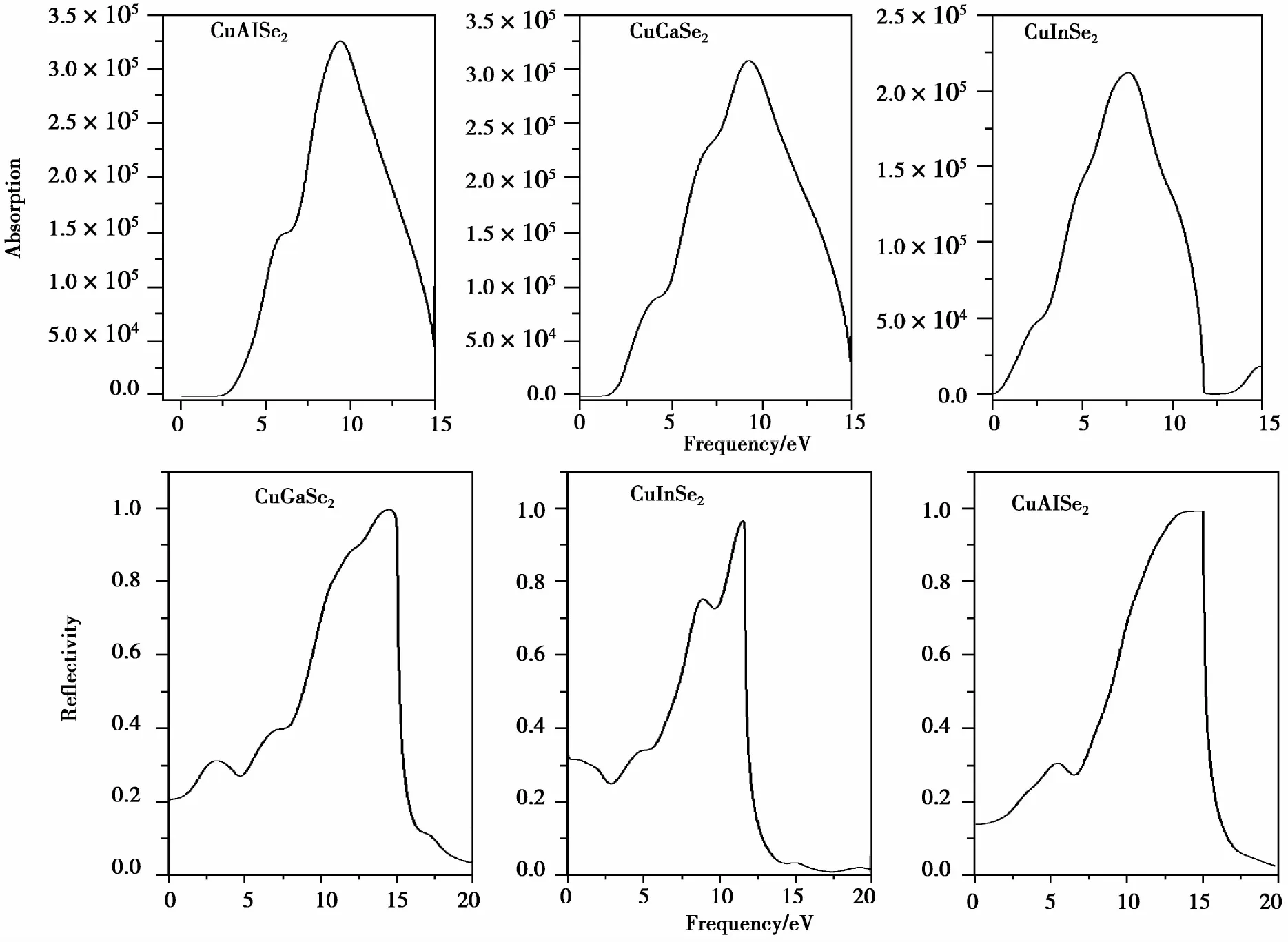
图4 CuXSe2(X=Al,Ga,In)晶体吸收系数和反射率变化曲线Fig.4 Variation curves of reflectivity (R) and adsorption coefficient (α) of CuXSe2(X=Al,Ga,In)crystals
图4为CuXSe2的吸收系数和反射率随能量的变化曲线。其中, 对于CuAlSe2和CuGaSe2, 它们曲线变化从整体上看相对于CuInSe2更为相似。在可见光区域(1.62~3.11 eV)的反射率随着能量增加,在3.11 eV处分别为22%和31%。而CuInSe2的反射率在可见光区域是有所减小的,在2.86 eV处达到最小为25%。对吸收系数变化曲线而言,3种材料在可见区域都随能量增加开始增加, CuAlSe2变化曲线较陡,CuGaSe2和CuInSe2变化曲线相对缓慢一些。在弱吸收区域(0 eV附近),CuAlSe2区域范围为0~2.63 eV, CuGaSe2区域范围为0~1.70 eV,和带隙的值吻合很好,但是CuInSe2的区域却为0~0.34 eV,与带隙的值相差较大,产生这个结果的原因在文献[18]中有描述。CuXSe2(X=Al,Ga,In)3种材料的吸收峰分别是紫外区的9.52,9.38,7.52 eV。要想使其成为良好的光吸收材料,还需通过金属掺杂等方法将其进一步调整光吸收区。
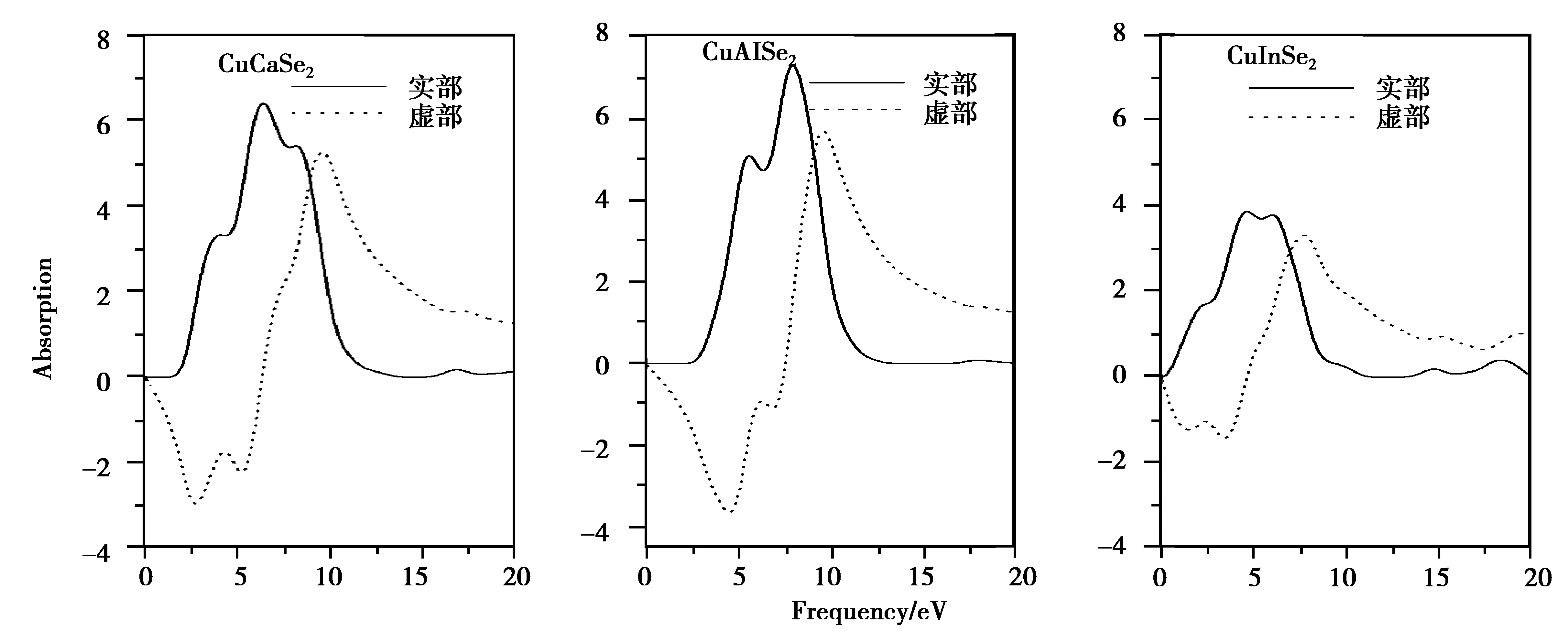
图5 CuXSe2(X=Al,Ga,In)晶体光电导率实部和虚部变化曲线Fig.5 Variation curves of real and imaginary parts of photoconductivity of CuXSe2(X=Al,Ga,In)crystals
在铜基半导体材料系里晶体中Se2(简称CIS)是一种非常重要的且研究最多的太阳能电池材料[18-21],并且已基本实现了商业化。为此可对相同结构的CuXSe2(X=Al,Ga,In)光电性质进行研究和对比。图5给出了3种材料光电导率实部和虚部的变化情况, 并对计算结果加以比较。由图5可看出在低能量区域CuAlSe2和CuGaSe2与CuInSe2有明显的平缓区域,而CuInSe2的光电导率会随着光子能量增加而增大。在随后的能量变化区域三类晶体电导率曲线的峰形比较相似,呈现出了双峰结构,且三类材料的最大值都在紫外波段。半导体材料能不能成为较好的太阳能电池,要求有较强的吸收能力和较小的反射率。而带隙较小的材料对太阳光吸收的更多。在能量比带隙区域小的太阳光是不会被吸收的,同时此区域的光电导率也为零。而在CuXSe2(X=Al,Ga,In)三类材料中,CuInSe2和CuGaSe2带隙都比CuAlSe2小得多,能较好的吸收太阳光,因此说明CuAlSe2并不适合做太阳能材料,而CuGaSe2却有很好的太阳能材料的潜质。
4 总 结
采用密度泛函理论框架下的平面波赝势,应用第一性原理方法对二元半导体ZnSe和三元半导体CuXSe2(X=Al,Ga,In)的电子对结构的影响进行了研究,计算得到了两类半导体的晶格常数、化学键键长。重点研究了三元半导体的能带结构和光学性质并进行分析而得到结论:
(1) 二元半导体ZnSe和三元半导体CuXSe2(X=Al,Ga,In)是直接带隙半导体材料,其晶格参数与实验值符合很好,且在CuXSe2系列晶体中按照Al→Ga→In的顺序增加。
(2) 由三元半导体能带图看出,带隙按照Al→Ga→In依次减小,在费米能附近主要由Cu原子的d电子轨道构成,且3d在此时峰值最大。
(3) 根据光学性质研究发现,在三元半导体材料中平均介电函数在零频时按照Al→Ga→In依次增大;吸收系数最强峰值都出现在紫外区域,可通过掺杂金属等方法进行改进。光电导率曲线对比发现,在CuXSe2(X=Al,Ga,In)3种半导体材料中,CuInSe2是适合做太阳能光电材料的,而CuGaSe2比CuAlSe2有更好的太阳能材料潜质。
[1] 刘芳芳,何清.Cu(In,Ga)Se2材料对其电池性能的影响[J].半导体学报,2005,26(10):1954-1958
LIU F F,HE Q.The Influence of a CIGS thin Film Composition on Performance of a Solar Cell[J].Chinese Journal of Semiconductors,2005,26(10):1954-1958
[2] MIGUEL A,CONTRERAS K,RAMANATHAN J,et al.Diode Characteristics in State-of-the-art ZnO/CdS/Cu(In1-xGax)Se2Solar Cells[R].Prog Photovolt:Res APPl,2005
[3] CHAURE N B,YOUNGE J,SAMANTILLEKE A P,et al.Electrodeposition of P-i-n Type CulnSe2Nutilayers for Photovoltaic Applications[J].Solar Energy Material & Solar Cells,2004,81:125-133
[4] JASENEK A.Electrodeposition of CulnSe2thin Films and Their Characteristics[J].Physica B,1999,266:192-197
[5] NAKAMURA S,SUGAWAR A,HASHIMOTO A,et al.Composition Control of Electrode Posited Cu-In-Se Layers for Thin Film CulnSe2Preparation[J].Solar Energy Material & Solar Cells,1998,50:25-30[6] TZVETKOV A E,NSTRATIEVA,MGANEHE V,et al.Preparation and Structure of Annealed CulnSe2Electrodeposited Films[J].Thin Solid Films,1997,311:101-106
[7] STRATIEVA N,TZVETKOVA E,MGANEHE V,et al.Structural and Optical Properties of Electrodeposited CulnSe2Layers[J].Solar Energy Material & Solar Cells,1997,45:87-96
[8] JASENEK A,RAU U,WWINERT K,et al.Illumination-induced Recovery of Cu(In,Ga)Se2Solar Cells after High-energy Eleetron Irradiation[J].APPI Phys Lett,2003,82(9):1410-1412
[9] KUNDU S N,BASU M,CHAUDHURI S,et al.CulnSe2Films Produced by Graphite Box Annealing of Multilayer Precursors[J].Thin Solid Films,1999,339:44-50
[10] CHEN S,GONG X G,WALSH A,et al.Electronic Structure and Stability of Quaternary Chalcogenide Semiconductors Derived from Cation Cross-substitution of II-VI and I-III-VI2Compounds[J].Phys Rev B,2009,79(16):165211
[11] PERDEW J P,BURKE K,EMZERHOF M.Generalized Gradient Approximationmade Simple[J].Phys Rev Lett,1996,77(18):3865-3868
[12] MONKHORST H J,PACK J D.Special Points for Brillouin-zone Integrations[J].Phys Rev B,1976,13(12):5188-5192
[13] PACK J D,MONKHORST H J.“Special points for brillouin-zone integra-tions”-A reply[J].Phys Rev B,1977,16(4):1748-1749
[14] STAMPFL C,VAN-DE-WALLE C G.Density-functional Calculations for III-V Nitrides Using the Local-density Approximation and the Generalized Gradient Approximation[J].Phys Rev B,1999,59(8):5521-5535
[15] JAFFE J E,ALEX Z.Theory of the Band-gap Anomaly in ABC(2) Chalcopyrite Semiconductors [J].Phys Rev B,1984,29(4):1882-1906
[16] SONALI S,SINHA T P,ABHIJIT M.Electronic Structure Chemical Bonding and Optical Properties of Paraelectric BaTiO3[J].Phys Rev B,2000,62(12),8828-8834
[17] 周和根,黄铜矿型半导体材料CuAlX2(X=S,Se,Te)的电子结构和光学性质[J].物理化学学报,2011,27 (12),2805-2813
ZHOU H G.Electronic Structures and Optical Properties of CuAlX2(X=S,Se,Te) Semiconductors with a Chalcopyrite Structure[J].Acta Phys Chim Sin,2011,27 (12),2805-2813
[18] 冯晶,肖冰,陈敬超,CuInSe2电子结构与光学性质的第一性原理计算[J].物理学报,2007,56(10),5990-5995
FENG J,XIAO B,CHEN J C.Electronic and Optical Properties of CuInSe2from Ab-initio Calculations[J].Acta Physica Sinica,2007,56(10):5990-5995
[19] CHICHIBU S F,OHMORI T,SHIBATA N.Fabrication of P-CuGaS2/n-ZnO:Al Heterojunction Light-emitting Diode Grown by Metalorganic Vapor Phase Epitaxy and Helicon-wave-excited-plasma Sputtering Methods[J].J Phys Chem Solids,2005,66,1868-1871
[20] MITCHELL K W,EBERSPACHER C,EEMER J H.CuInSe2Cells and Modules[J].IEEE Transacation on Electron Device 1990,37(2),410-417
[21] SHIMIZU A,CHAISITSAK S,SUGIYAMA T.Zinc-based Buffer Layer in the Cu(InGa)Se2Thin Film Solar Cells [J].Thin Solid Films,2000,361-362,193-197
责任编辑:田 静
Study on the Structure and Optical Properties of ZnSe and CuXSe2(X=Al, Ga, In)
DONG Yu-jing1,GAO Yan-li1
(School of Science and Technology, Xinyang University, Henan Xinyang 464000, China)
The crystal structure of solar material ZnSe and CuXSe2(X=Al.Ga, in) has been optimized firstly by using the pseudopotential plane wave basis set of density functional theory, studied the lattice parameters, bond lengths, and forecasted the CuXSe2band gap and optical properties. The results indicate that the band gap decreased in accordance with Al→Ga→In, but the lattice parameters and deformation parameters increased.The optical properties of dielectric function, absorption coefficient, reflectivity and photoconductive rate have been analised and foud that the strongest peaks of the absorption coefficient are in the ultraviolet region. The optical properties increased in accordance with Al→Ga→In successively in three crystals.
density functional method; semiconductor; band gap; optical property
2016-08-11;
2016-10-12.
河南省高等学校重点科研项目计划(15A140037)
董玉静(1984-),女,河南新乡市人,讲师,硕士,从事半导体材料的理论研究。
10.16055/j.issn.1672-058X.2017.0002.017
TM914.4
A
1672-058X(2017)02-0084-06
猜你喜欢
中学生数理化·高一版(2022年3期)2022-04-15
数学学习与研究(2020年23期)2020-01-11
制冷(2019年2期)2019-12-09
电镀与环保(2016年4期)2017-01-20
电镀与环保(2016年3期)2017-01-20
卷宗(2016年8期)2016-11-15
光学精密工程(2016年5期)2016-11-07
浙江大学学报(工学版)(2016年2期)2016-06-05
电测与仪表(2016年2期)2016-04-12
中国惯性技术学报(2015年1期)2015-12-19
