
栽培种花生ddGBS测序中酶切组合的选择
2017-03-20 00:25李春娟闫彩霞赵小波单世华
花生学报 2017年4期
王 娟,李春娟,闫彩霞,赵小波,单世华
(山东省花生研究所,山东 青岛 266100)
花生(ArachishypogaeaL.)是我国重要的经济作物和油料作物,我国的花生产量占世界花生总产的40%以上,居世界第一位[1]。栽培种花生基因组(AABB)庞大而复杂[2],导致对花生基因组的测序和分析非常困难。为解决类似难题,科学家们基于只对非重复或低重复基因组区域进行测序来降低测序基因组复杂程度的简化基因组测序(Reduced-Representation Genome Sequencing, RRGS),开发了一系列低成本、高通量的基因型鉴定方法。其中,基于测序的基因分型(Genotyping by Sequencing,GBS)方法是通过获取全基因组范围内呈现特异性酶切位点附近的小片段DNA标签,以获得整个基因组的序列特征,从而进行全基因组水平的生物信息学分析[3]。随着测序技术的不断改进,双酶切GBS(Double digest Genotyping-by Sequencing, ddGBS)测序能够降低基因组复杂程度,使得全基因组水平的基因分型更加简便、可靠、实用[4]。
因此,ddGBS能够为进一步揭示花生种质资源中的新基因,以及确定影响花生生长发育的关键基因构建理论基础。作为GBS测序的第一步,对限制性内切酶组合的选择决定了酶切位点附近的小片段DNA分布,对于后续基因信息的识别有重要的影响。本研究对三组常用的酶切组合进行比较[5-6],选取了最佳酶切组合,并且通过统计分析进一步确认该酶切组合的合理性。
1 材料与方法
1.1 花生全基因组序列的获取
本研究所用参考基因组序列来自花生数据库(https://www.peanutbase.org/)。其中,Arachisduranensis的基因组(A基因组)大小为1.08G,A.ipaensis的基因组 (B基因组)大小为1.35G。
1.2 三个候选酶切组合的比较及统计分析
根据文献报道,选取三组常用的限制性内切酶组合(SacI和MseI;PstI和MspI;EcoRI和NIaIII)。其中,SacI的识别位点为GAGCT^C,MseI的识别位点为T^TAA,PstI的识别位点是CTGCA^G和G^ACGTC,MspI的识别位点为C^CGG,EcoRI的识别位点为G^AATTC,NIaIII的识别位点是CATG。选择依据:① 酶切片段在各染色体上分布较均匀;② 酶切片段在全基因组上覆盖度较高。通过电子酶切统计结果,初步选定酶切组合[7]。
通过R软件包(https://www.r-project.org/)和Excel软件统计候选酶切组合得到酶切片段长度。通过R软件包统计该酶切组合在全基因组和各个染色体上的分布、数目和长度。
2 结果与分析
2.1 最佳酶切组合的选取
针对群体分析的研究目的,测序深度选择在5~10X左右。依次使用三组常用的限制性内切酶组合(SacI和MseI;PstI和MspI;EcoRI和NIaIII)对已公布的花生野生二倍体基因组A(Arachisduranensis)和基因组B(A.ipaensis)序列进行电子酶切。结果显示,EcoRI和NIaIII酶切组合得到的酶切片段覆盖全基因组比例最大,A基因组上占3.10%,B基因组上占3.53%;PstI和MspI得到的酶切片段覆盖全基因组范围最小,A基因组上占0.88%,B基因组上占1.19%。因此,EcoRI和NIaIII酶切组合成为候选酶切组合,相应测序量在403~806 M之间(表1)。
2.2 酶切片段的相关统计
EcoRI和NIaIII酶切组合所产生酶切片段长度如下(图1)。基因组A和B上,大小在1~10kb片段所占比例最大,>40kb以上的片段占比例最小。高通量测序通常选取大小在300~500 bp酶切片段,本研究中,实际统计的有效长度在386~390 bp之间,片段数目在B05染色体上最多,达到27568个,A01染色体上最少,有7888个。有效Tags总数与染色体长度相关,所占染色体的比例稳定,除A08染色体为0.15外,均为0.14(表2)。
在花生全基因组范围内,有效tags总数为412662个,花生各个染色体上的分布位置见图2,酶切片段分布较均匀,有效覆盖度2.46%(表3),能够达到GBS测序所需的覆盖范围(1%~3%),因此,酶切所得到DNA片段的覆盖范围对于后续的分析也比较合理。
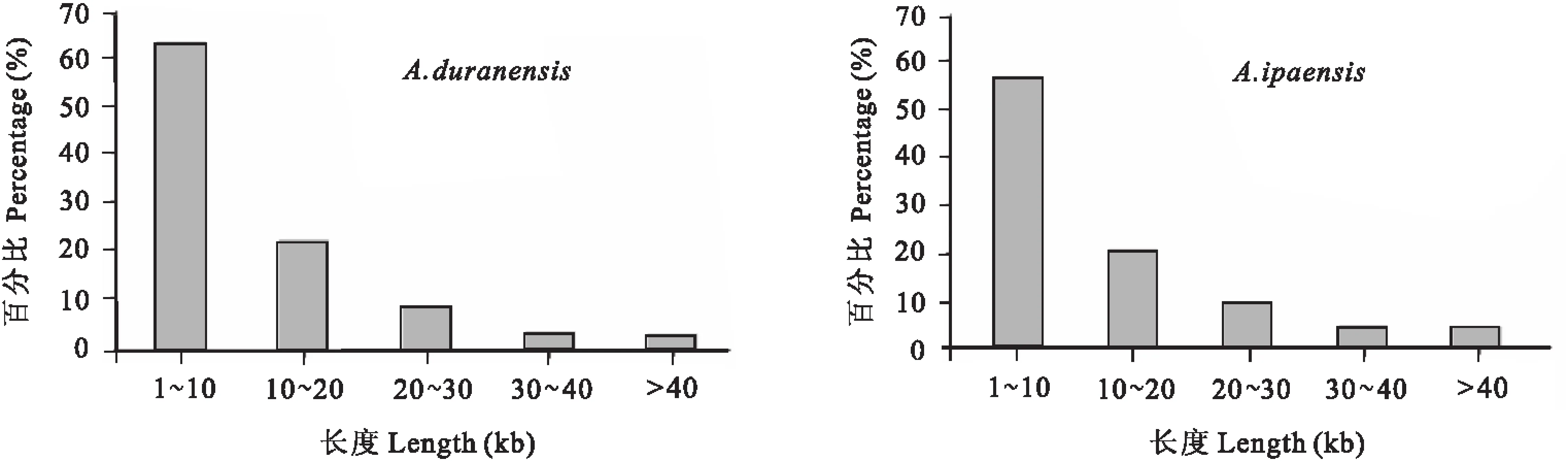
图1 酶切片段长度柱形统计图 Fig.1 The bar chart of the enzyme fragment length
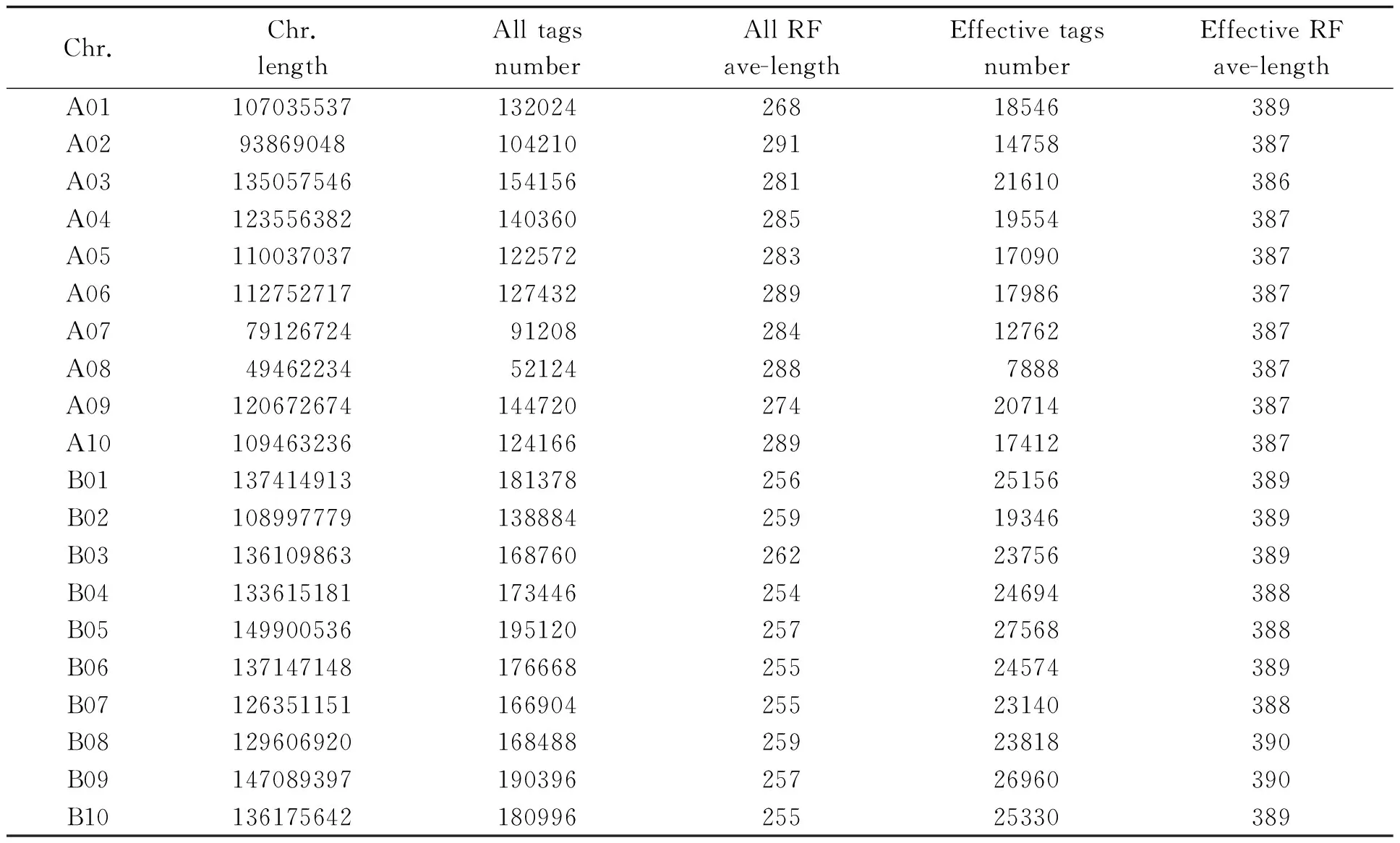
表2 酶切片段在每条染色体上的分布情况
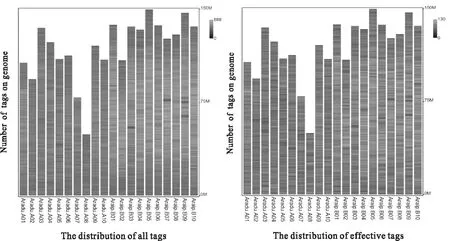
图2 酶切片段分布(左)以及有效酶切片段的分布(右) Fig.2 The distribution of restriction fragments (left) and the effective of restriction fragments (right)

表3 酶切片段在全基因组上的分布情况
3 讨论与结论
GBS是一种性价比较高的简化基因组测序方法。GBS测序首先需要选择最佳的酶切组合来保证后续测序和分析的正常进行。根据参考文献信息,研究选取了三组常用的限制性内切酶组合(SacI和MseI;PstI和MspI;EcoRI和NIaIII),并分别对已公布的花生野生二倍体基因组A(Arachisduranensis)和基因组B(Arachisipaensis)序列进行电子酶切和相关统计。
通过对酶切片段覆盖范围,数目以及长度统计数据进行分析和对比,表明EcoRI和NIaIII是相对比较理想的限制性内切酶组合,因此本研究为群体遗传资源在全基因组水平的挖掘奠定了基础。
[1] 万书波. 中国花生品质区划[M]. 北京:科学出版社,2012.
[2] 禹山林.中国花生品种及其系谱[M]. 上海:上海科学技术出版社, 2008.
[3] Davey J W, Hohenlohe P A, Etter P D, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing [J]. Nature Reviews Genetics, 2011, 12(7): 499-510.
[4] Elshire R J, Glaubitz J C, Sun Q, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species [J]. PloS ONE, 2011, 6: e19379. DOI:10.1371/journal.pone.0019379.
[5] Shirasawa K, Hirakawa H, and Isobe S. Analytical workflow of double-digest restriction site-associated DNA sequencing based on empirical and in silico optimization in tomato [J]. DNA Research, 2016, 23:145-153.
[6] Zhou X, Xia Y, Ren X, et al. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq) [J]. BMC Genomics, 2014, 15(1):351.
[7] Bertioli D J, Cannon S B, Froenicke L, et al. The genome sequences ofArachisduranensisandArachisipaensis, the diploid ancestors of cultivated peanut [J]. Nature Genetics, 2016, 48(4):118-120.
猜你喜欢
能源与环保(2022年3期)2022-04-08
中学生数理化·中考版(2021年9期)2021-11-20
水利技术监督(2021年1期)2021-01-21
科学与财富(2020年15期)2020-07-04
移动通信(2020年4期)2020-05-07
中国港湾建设(2019年12期)2019-12-18
天津城建大学学报(2019年5期)2019-11-07
移动通信(2019年4期)2019-06-25
中国当代医药(2015年17期)2015-03-01
中学生天地·高中学习版(2008年1期)2008-02-19
