
α-Fe2O3光电催化分解水制备氢气研究进展
2017-02-17 12:53王开放刘光高旭升贺冬莹李晋平
化工进展 2017年2期
王开放,刘光,高旭升,贺冬莹,李晋平
(太原理工大学精细化工研究所,山西 太原 030024)
α-Fe2O3光电催化分解水制备氢气研究进展
王开放,刘光,高旭升,贺冬莹,李晋平
(太原理工大学精细化工研究所,山西 太原 030024)
光电化学池可以将太阳能以氢气的形式储存起来,其中稳定、廉价的催化剂是关键。α-Fe2O3具有合适的禁带宽度,较高的理论光-电转化效率,光稳定性好,在地壳中的储量丰富,被认为是最具有发展前景的光电催化材料之一;但是它的导电性差、光生电荷寿命短、氧化反应过电位高,严重阻碍了其发展。本文首先介绍了光电催化理论,然后重点综述了近些年α-Fe2O3纳米结构的制备技术,以及针对其不足所采用的改性方法,包括通过元素掺杂来增强α-Fe2O3的导电性,表面处理来降低氧化反应过电势或陷阱浓度,与其他材料复合来增加光生电压或催化剂表面积,最后对α-Fe2O3作为光阳极催化剂分解水制氢未来的发展前景作出展望,指出多种手段的有效结合是提高其光电流密度的重要途径。
赤铁矿;太阳能;光电催化;水解;氢气
随着全球经济的不断发展,人类对能源的需求量持续扩大,全球能源的消耗仍然是以化石能源为主,但是化石燃料储量有限、生成周期长,难以满足持续大量的需求,而且常规能源的广泛应用所引起的环境问题日益凸显。解决能源问题的根本途径是寻找可替代能源。发达国家和发展中国家都投入了大量的资金、技术开发新的能源,包括太阳能、风能、地热能、生物质能等,这些新能源的普遍特点是污染小、储量大、可再生;其中太阳能被认为是最具有开发前景的能源之一,直接来源于太阳的辐射,源源不断地为地球提供能量,但是在地球上固定的地域接受的太阳能受到时间、气候和周围环境等影响,如何连续稳定地利用太阳能也是关注的方向之一。
自然界中绿色植物可利用太阳光将CO2和H2O转化为有机物储存起来,所以可以模拟光合作用,把太阳能转化为易于存储的形式。氢气是一种极为优越的能源,它的能量密度高(143kJ/g),燃烧产物为水,而且可以由水分解制取。1972年,科学家发现TiO2在光照下可以分解水为O2和H2,这一现象迅速引起了全球的关注,经过四十多年的发展,研究者们陆续发现多种物质具有光催化分解水的作用,这说明光电催化是具有发展潜力的方向之一。在这些新发现的材料中,有ZnO、WO3、BiVO4、CdS、α-Fe2O3等,吸收太阳光的光谱范围从最初的紫外光波段拓展到可见光区,提高了对太阳光的利用率,如图1。
α-Fe2O3又称作赤铁矿(hematite),有类似于刚玉的晶体结构,是铁的氧化物中相对稳定的一种物质,呈现弱磁性,广泛存在于自然界中。晶体中有两种不同长度的Fe—O键,(001)基面有相对较强的导电性[2]。1976年,HARDEE等[3]第一次发现α-Fe2O3具有光电分解水的性质,它具有合适的禁带宽度(1.9~2.2eV)[4],可吸收约40%的太阳光;当禁带宽度为2.03eV(600nm)时,光能转化为氢气的理论效率为16.8%[4];光稳定性好,能在pH>4的电解液中进行光电催化反应[5]。但是与其他光催化剂类似,α-Fe2O3有其固有的缺陷:单相的α-Fe2O3被认为是电的不良导体,使得光生电子和空穴的快速转移受到阻碍,是造成两种载流子易于复合的原因之一;与TiO2相比,α-Fe2O3的空穴扩散长度(LD)仅为1.5~5nm[6];此外,其氧化分解水过电位高,需要在较高的偏压下才能产生足够强的光电流。
近年来,研究者们运用多种技术手段对α-Fe2O3进行表征,尝试阐述产生上述问题的深层原因。本文综述了近些年国内外的研究成果,从α-Fe2O3光电水解制氢理论基础、制备技术、表征手段以及对它的改性手段,如元素掺杂、表面处理、复合材料方面分别作概述。
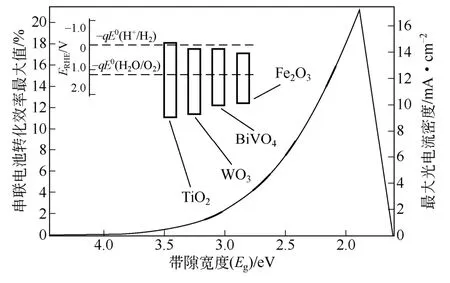
图1 常见光阳极半导体氧化物的光-电、光-氢的转化效率和带隙范围[1]
1 理论基础
α-Fe2O3光电催化分解水理论主要涉及两个方面:一是需要构建光电化学池(PEC);二是需要太阳光对催化剂的价带进行电子激发。
1.1 光电化学池
在电化学中,水的分解涉及两个氧化还原对O2/H2O(1.23Vvs. NHE)和H+/H2O(0Vvs.NHE),因此,实现水分解为H2和O2的最小电压为1.23V。理想的光催化剂的带隙应该横跨分解水的氧化电势和还原电势,即光催化剂能带(VB)的位置处于氧化电势(O2/H2O)的上方,导带(CB)位置处于还原电势(H+/H2O)的下方;能带和导带的位置不能向两侧偏离得太多,否则会导致带隙较大,只能利用太阳光中能量较高的那部分光能,降低了对太阳光的利用率。总之,能带和导带的位置既要满足分解水的需要,又不能过宽(小于3.0eV)。上述条件对催化剂提出了苛刻的要求,难以同时满足。α-Fe2O3的价带顶端比氧化电势高,同时导带底端也比还原电势高,造成其带隙并没有横跨氧化和还原电势,只是部分重叠,所以α-Fe2O3只适宜做光阳极催化剂,这就需要构建光电化学池(PEC)。在光电化学池装置里,α-Fe2O3在阳极作为催化剂,另一种材料一般为贵金属Pt,在阴极作为析氢催化剂,这时仍需要外加电压使得阴极的费米能级高于析氢电势产生氢气。阳极氧化反应是一个四电子的转移过程,实际上能带的位置需要比氧化电势高出许多,如图2。
1.2 能带激发
在半导体中,价带顶和导带底之间存在的能量差值称为禁带宽度,这是产生电子激发所需要的最小能量,它决定半导体被激发的光谱范围。α-Fe2O3的禁带是在氧元素2p能带和铁元素3d能带间形成的[8]。光照下价带顶的价电子吸收足够的能量后,跨过禁带,跃迁至导带底的空轨道上成为自由电子,在能带上则留下空穴(正电荷),如果价电子吸收的能量更多,可以从能带顶附近更低的能级向导带底附近更高的能级跃迁,产生更多的空穴和电子。空穴在α-Fe2O3中以极化子跳跃的方式迁移至催化剂的表面[9],发生水的氧化反应,电子通过PEC中集流体的收集流向阴极,参与水的还原反应。
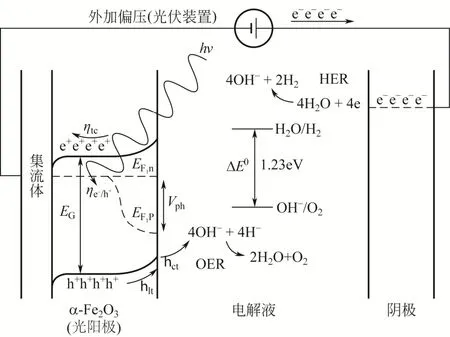
图2 光电化学池α-Fe2O3作为光阳极的能带示意图[7]
2 制备技术
在α-Fe2O3中,空穴的转移是以极化子的形式进行,但是由于空穴扩散长度短,容易与电子复合,缩小材料的微观尺寸则可以增大其相对扩散长度和与电解液的接触面积。α-Fe2O3微观形貌的纳米化可以用多种制备技术进行调控,这些纳米结构对其光电催化性能有重要的影响。
2.1 水热法
水热法是材料合成中常用的一种方法,它是将各种反应物均匀地混合在一起,在特定的温度下反应一定的时间,利用此方法可以合成出α-Fe2O3的前体FeOOH,然后在高温下焙烧生成α-Fe2O3纳米颗粒,不同种类的无机盐和反应温度可以制备出多种形貌的纳米结构。水热法得到的α-Fe2O3纳米颗粒一般不能直接作为分解水的催化剂,通常是将其水热沉积到镀有氟掺杂氧化锡的玻璃上(FTO),利用表面的导电层进行电子传输,生长在FTO表面的纳米棒与基底结合牢固,可以增加α-Fe2O3与基底的导电性。BEERMANN等[11]用水热法制备出直径为5nm的α-Fe2O3纳米棒阵列,发现这种棒状结构有利于电子和空穴的传导,膜的厚度(纳米棒的平均长度)是影响光电催化性能的关键因素。热处理时的温度一般需要大于400℃[12],才能使前体充分地转化为α-Fe2O3晶型;过高的焙烧温度可以使纳米棒一定程度地熔融,有效降低表面的陷阱浓度,增加晶体的结晶性[13]和催化剂表面的亲水性[14],但是同时它也会降低FTO的导电性,所以过高温度的热处理时间较短。
2.2 常压化学气相沉积
常压化学气相沉积(APCVD)是一种比较成熟的技术,在实验室中和工业上都被广泛的应用。首先,反应物被加热成气相,在载气(惰性气体)的流动作用下,气相反应物被运输到加热的基片表面,然后,反应物沉积在高温的基片表面,并发生化学反应,调节载气流速和沉积时间,可以得到不同厚度的反应产物。α-Fe2O3第一次被用作光电催化反应运用的就是这种技术,得到的是n型α-Fe2O3。KAY等[15]运用这种方法得到花菜状(树枝状)的纳米结构,如图3。这种纳米尺度的结构表面是由尖锐的纳米颗粒堆积而成,具有很高的粗糙度,它们增大了与电解液的接触面积,使得电压降集中在纹理边界与电解液的界面处,促进在空间电荷层中吸收的光谱转化为电流,而且表面尖锐的纹理抑制了光生电荷在水平方向上的扩散,使得电荷只在垂直方向上传递,降低了光生电子-空穴复合的概率[16]。

图3 FTO上用常压化学气相沉积法得到的高分辨率Fe2O3薄膜[15]
2.3 原子层沉积
原子层沉积法(ALD)原理与化学气相沉积类似,在极低的压力下,对膜的控制更加精细,可以进行精确的维度控制[17]。用这种方法可以在合金TiSi2纳米网表面生成一层约25nm厚的α-Fe2O3层,如图4[18],在无外加助析氧催化剂的情况下,可以获得很高的光电流密度;而且还可以在催化剂的表面沉积钝化层,构建异质结构,负载助催化剂以及进行元素掺杂和改变催化剂的表面结构[19]。

图4 TiSi2和α-Fe2O3核/壳异质结构的低倍电子透射图[18]
2.4 喷雾高温分解法
喷雾高温分解技术(spray pyrolysis)则是将含铁的溶液直接以雾状的形式喷到高温的基片上,氧化生成α-Fe2O3,其中有多种因素会影响到催化剂的光电催化性能,包括铁源、溶剂、喷雾时间、基片温度等[20];通过调节喷雾的时间来制备不同厚度的薄膜,发现膜的厚度会对带隙产生轻微的改变,而且较薄的膜有利于电子-空穴的分离。
2.5 电沉积法
运用循环伏安法可以在FTO上沉积得到无定形的三价铁氢氧复合物膜,高温下焙烧,可以转化为α-Fe2O3,电沉积的电压、时间和热处理的温度、时间都影响到α-Fe2O3的形貌[21]。在弱酸性电解液中,电压为1.2V时,γ-FeOOH可以直接在阳极电沉积析出[22],热处理后转化为α-Fe2O3;而在无水的电解液中,也可电沉积得到含铁的前体,并且较高的沉积电流密度下可产生更小的晶粒和(110)晶面的优先生长,这对α-Fe2O3的光电催化性能是有利的。随后,SHINDE等[23]运用反向脉冲电沉积的方法制备出了多层纳米瓣结构的α-Fe2O3,与花菜状的结构类似,增加了催化剂与电解液的接触面积,外加电压为1.23V(RHE)时,其电流密度比常规电沉积方法得到的催化剂电流密度提高了1.8倍。在电沉积的过程中,运用模板辅助的方法可以制备纳米管结构的α-Fe2O3[24],其比纳米棒形貌有更高的光电化学活性。
2.6 铁箔氧化法
在特定的电解质溶液中,恒电势下可将铁箔氧化成多孔的α-Fe2O3,孔径为50~250nm,深度为300~600nm,这种多孔结构缩短了空穴向表面的扩散距离[25];在一定流速的氧气下,还可以在铁箔表面高温氧化得到α-Fe2O3纳米带状结构[26]。
2.7 溶胶法
溶胶法是将固体纳米颗粒制备成悬浮液,然后均匀地分散在电极上,有时需要高温处理来增强基底与材料之间的接触。这种方法获得的催化剂纳米颗粒随机地分散在电极表面,造成陷阱位点较多,易于成为电子空穴的复合中心,降低了光-电转化效率[27]。
在制备α-Fe2O3应用较多的几种方法中:电沉积的方法可以方便地调节膜的厚度;溶剂热法简单易行,可以制备出纳米棒、纳米管[28]、纳米球[29]、纳米花状[30]等结构,尤其是垂直生长在FTO上的纳米棒相对于平面的薄膜更加有利于电荷的汇集和传输,但它在相同条件下所获得的电流密度并不高;喷雾高温分解技术所获得的膜厚度难以均一,电沉积法可以比其获得更好的结晶性和多孔性,而且具有更低的平带电势;化学气相沉积和原子层沉积技术是两种有效的方法,尤其是后者可以进行精确的形貌调控,这是其他技术难以相比的,但是它的沉积速率慢,对设备和试剂的要求也较高。
3 性能表征
表征α-Fe2O3光电催化性能的方法有多种,主要有:表示外加电压与光电流密度关系的线性扫描伏安法(LSV)、测试光电流稳定性或光响应的计时电流法(chronoamperometry)、光-电转化效率(IPCE)和光能转化为氢能的效率(APCE),为了解释这些宏观的性能,需要进一步做一些电化学表征,如莫特-肖特基曲线(Mott-Schotty)、电化学阻抗(EIS)等。
通常情况下,α-Fe2O3的光电性能测试是在三电极结构的碱性电解液中进行的,外加偏压的作用不仅是促使阴极产生氢气,它还延长了光生空穴的寿命[31],不同种类和浓度的电解液pH不同,因此,为了便于比较,常将相对于参比电极的外加偏压转化为相对于可逆氢电极(RHE),具体公式为式(1)。

式中,E为相对于可逆氢电极的电势,V;Eapp为外加偏压,V;pH为电解液的酸度值;E0为参比电极电势,V。
光源一般为模拟一倍的太阳光(AM1.5G),其光照强度是100mW/cm2。SEGEV等[32]研究了不同的光照强度对α-Fe2O3性能的影响,发现光电流密度与光照强度呈线性关系,指出提高光照强度比增加外加电压更有利于电荷的转移。
3.1 光-电转化效率和光-氢转化效率
光-电转化效率可用式(2)表示。

式中,I为光电流密度,mA/cm2;P为光照强度,mW/cm2;λ为波长,nm。
光-氢转化效率计算如式(3)。

式中,I为光电流密度,mA/cm2;P为光照强度,mW/cm2;Eapp为外加偏压,V。
APCE和IPCE的关系[33]如式(4)。

式中,A为电极的吸光率。
3.2 带隙计算
通过测量α-Fe2O3吸收光谱波长的范围,可以计算带隙值Eg(band gap)[11],如式(5)。


式中,λ为α-Fe2O3能吸收光谱的最大波长,nm。
3.3 莫特-肖特基曲线
莫特-肖特基曲线常被用于研究金属氧化物半导体的光电性能,其应用基于两个假设:在半导体与电解液的界面存在多种电容,如空间电荷电容层、亥姆霍兹电容层、表面电容层等,这里只考虑空间电荷电容层,其余的忽略;需要构建一个由多种电阻和电容组成的等效电路,测试频率可以为几千赫兹,如式(7)。
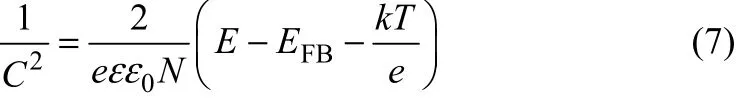
式中,C为单位表面积空间电荷电容,F/cm2;E为元电荷,1.60×10–19C;ε为α-Fe2O3的相对绝缘常数[34];ε0为真空介电常数,8.85×10–12F/m;N为n型半导体供体的浓度ND,p型半导体空穴的浓度NA,cm–3;E为外加偏压,V;EFB为平带电势,V;k为玻兹曼常数,1.38×10–23J/K;T为绝对温度,K。
常温下,kT/e约为25mV,可忽略不计。测试时,为了尽可能减少频率对莫特-肖特基曲线(M-S)的弥散影响,一般选择1000Hz,而且不宜应用于粗糙度较大的电极(如多孔表面)。当曲线的斜率为正值时,电极为n型半导体;当曲线的斜率为负值时,电极则为p型半导体。曲线在横轴上(电压轴)的截距为平带电势(EFB),较低的平带电势可以抑制光生电子和空穴的复合,有利于开口电压的降低;供体的浓度可以用曲线的斜率(slop)计算,如式(8)。

ND浓度的大小反映出α-Fe2O3的导电性能,元素掺杂一般会使ND数值升高,不同的方法制备出的α-Fe2O3,ND也有变化,一般是在1016~1022cm–3[35-40]。α-Fe2O3与电解液接触时,电荷会从催化剂表面迁移至电解液中,导致平带电势增大,当对电极施加电压时,电压降主要击垮空间电荷区电容,会进一步影响到平带电势。
在电解液与电极的界面处,电荷的迁移会形成一个静电区域,在这个区域内,电荷载体都会被转移出去而形成一个电荷的消耗层,光生电子-空穴在此消耗层内被分离。消耗层的宽度(Wd)可用式(9)计算。

消耗层的厚度是随着外加偏压变化的,偏压的增加使Wd变宽,同时也会延长光生空穴的寿命,提高了电荷载体的分离效率,如图5。
3.4 电化学阻抗
电化学阻抗是以小振幅的正弦电势为扰动信号,研究电极在宽频率范围内的表面容抗,为此需要构建一个电极和电解液的等效电路,如图6。
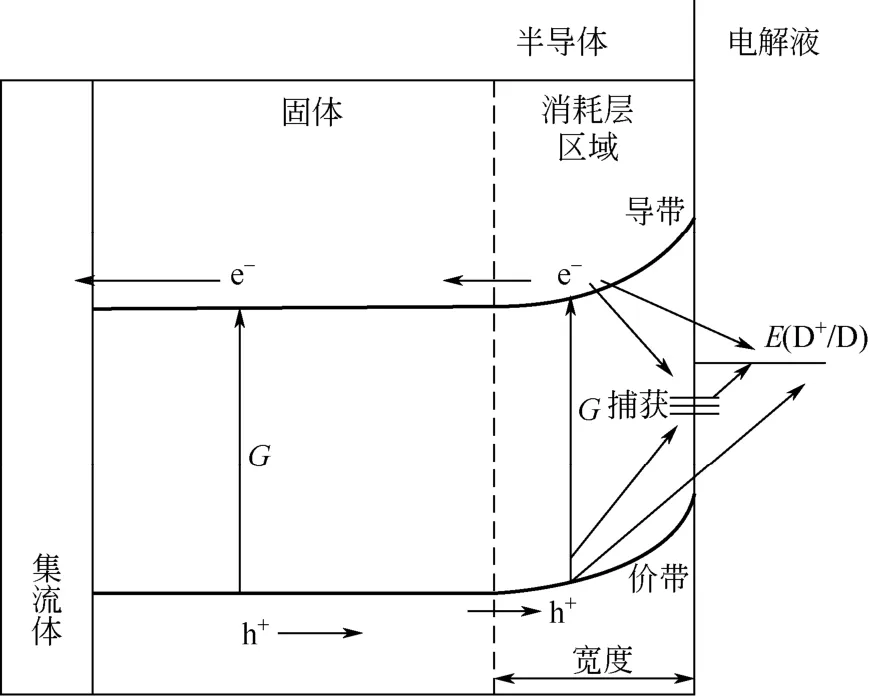
图5 在α-Fe2O3电极上电荷载体动力学的物理模型[41]

图6 等效电路的物理模型[42]
等效电路中一般包括电极和电解液的电阻(Rs)、电极表面电容(Cbulk)、价带中电荷转移电阻(Rct,bulk)、体积电容(Ctrap)、表面空穴复合电阻(Rtraping)、表面电荷转移电阻(Rct,trap)。影响电极表面状态的主要是价带中的电荷转移电阻和体积电容,电荷转移电阻越小,开口电流就越大,说明氧化分解水的过程发生在电极表面,而不是产生光生空穴的能带上[42]。提高外加偏压可以使体积电容减小,加速空穴的转移速度,这与M-S曲线中的电荷消耗层类似。
电化学阻抗谱反映的是电极总的电阻和电容结果,光照和外加偏压都会使总电容值降低,而对电阻影响不大,说明电极的电压降主要发生在电容层中。
4 改性技术
对α-Fe2O3的不足,目前文献中报道了多种方法对其进行改性,可以分为3类:一是元素的掺杂,主要是过渡金属元素和一些主族金属元素;二是表面处理,通过在半导体表面覆盖一些特殊的涂层,影响空穴在界面处参与氧化反应的状态;三是与其他材料复合,这样可以改善集流体对电子的收集状况,提高对光能的利用率。
4.1 元素掺杂
单相的α-Fe2O3是电的不良导体,离子掺杂是提高其导电性的有效手段之一,掺杂离子替代晶体中三价铁离子的位置,由于价态和离子半径的不同,会产生一定程度的晶格畸变。高价态的离子(大于+3价)在晶格中可以给出电子,形成n型半导体,使费米能级弯向导带的底端;低价态的离子(小于+3价)起到电子受体的作用,可以形成p型半导体,使费米能级弯向价带的顶端[26]。有一些元素的引入还会影响到α-Fe2O3的生成形貌。α-Fe2O3自身是n型半导体,在光照下产生光阳极电流,可以作为光阳极催化剂,但是这种性质并非固定不变,周围的气氛会影响此材料表面氧的吸附,发生能带弯曲,造成n-p型之间的转变[43]。
FTO常被用作α-Fe2O3的基底,在高温处理α-Fe2O3的前体时,锡元素不可避免地要向催化剂材料中扩散。LING等[44]研究了FTO表面锡元素在不同温度下对α-Fe2O3的掺杂情况,在大于650℃时随着温度的升高,锡元素的含量逐渐升高,800℃热处理后,在外加偏压1.23V(RHE)和一倍太阳光(AM1.5G)下其光电流密度为1.24mA/cm2,而550℃处理后的仅为0.035mA/cm2。这种利用高温扩散的方法,元素的掺杂量是不可控的,随后他们又将定量SnCl4的乙醇溶液加入到制备前体的溶液中,高温焙烧后由原来未掺杂时的棒状变成珊瑚状(图7),所获得的电流密度也提升至1.86mA/cm2。将SnCl4的乙醇溶液滴涂在α-Fe2O3纳米片上,在1000℃的火焰中短暂处理后快速降温,可以对α-Fe2O3纳米片进行表面锡掺杂和修饰,其光电流密度提升近一倍[45]。WANG等[46]用非极性的有机溶剂甲苯作为溶剂,分别以油酸铁和辛酸亚锡为铁源和锡源,制备出了385nm厚的α-Fe2O3薄膜,使得光生电流密度达到了3.32mA/cm2,这是目前仅用掺杂方法获得的最高值,并且表现出了良好的稳定性。

图7 未掺杂和锡掺杂的α-Fe2O3纳米线电镜扫描图[44]
镁的掺杂可使α-Fe2O3从n型光催化材料转化为p型[33],并且不改变它的结构、形貌以及光学性能,将此p型材料覆盖在单相n型α-Fe2O3上可形成p-n结,这种结构成一个内部的区域,可加速光生电子-空穴的分离,促进空穴向电解液中转移,从而降低了开口电压,这些作用可被电化学阻抗谱(EIS)证明。
水热合成α-Fe2O3的前体时,加入5%(摩尔分数)Mn2+,可使电流密度比未掺杂的提高75%,锰元素的加入可降低空穴转移的能量势垒,加速电子-空穴的分离[38]。用同样的方法引入一定量的Ge胶体溶液时,则制备出了约10nm厚的α-Fe2O3纳米片,使电流密度提高了50倍,并且发现退火温度是影响催化性能的主要因素之一[39]。
在电解液中加入Cr3+、Mo5+[47]、Cd3+[48]、Zr4+[36]、Ni2+[49],可与Fe3+共沉积,热处理后可以形成相应的元素掺杂,这些元素的掺杂都不同程度地提高了α-Fe2O3的导电性,并降低了其开口电压,但是各自最优的掺杂量是不同的。Mo、Ni并不是均匀地分布在α-Fe2O3中,在表面的含量较多。锆掺杂获得的供体浓度可达2.6×1021cm–3,这是其光电流密度较高的主要原因。镉元素的掺杂中出现了杂相CdO,这说明加入的镉元素有一部分是晶间掺杂,镉的引入也降低了α-Fe2O3的平带电势。
ZHANG等[50]用常压化学气相沉积法制备了钛掺杂的α-Fe2O3,不同的掺杂量对形貌的影响很大,而且钛的引入可使晶体沿着(110)晶面选择性生长,改变了晶体的结构;同样的方法,硅掺杂调控出了花菜状的纳米形貌。FRANKING等[51]用钛掺杂α-Fe2O3纳米线,可使供体的浓度提高10倍;钛的引入改变了催化剂表面的电子结构,起到了类似钝化的作用。
贵金属Pt是优异的催化剂,其被引入α-Fe2O3晶体中进行掺杂,产生了显著的影响,可使太阳光转化为氢气的效率达到5%[52]。在对其他材料的元素掺杂研究表明,非金属元素的引入可以调节催化剂的能级[53],一些金属元素还可以延缓光生电子-空穴的复合[54],对α-Fe2O3的掺杂具有指导意义。
应用密度泛函理论(DFT)中的局域密度近似法(LDA)模拟了Al掺杂对α-Fe2O3电子结构的影响:铝离子取代了铁离子的位置,增强了导电性,促进了电荷的极化跃迁,但是没有改变带隙边缘的静电场结构[55]。LIAO等[56]在密度泛函理论中加入参数U计算了对羟基化的α-Fe2O3表面掺杂钛、锰、钴、镍、氟、硅催化分解水的动力学反应;不同元素的掺杂可适当调节催化剂表面催化反应的中间产物所形成的键能,如果催化剂表面产生的缺陷有铁原子吸附和氧空缺时,更有利于分解水的反应,如图8。

图8 真空条件下α-Fe2O3表面水氧化反应最优结构的中间产物路径示意图[56]
4.2 表面处理
光电催化分解水生成氧气是一个发生在电解液与催化剂界面处的四电子转移反应,动力学[57]和有效的空穴浓度[42]是光电催化析氧反应的主要影响因素。α-Fe2O3表面处的析氧反应动力学迟钝,缺陷较多,易成为电子-空穴的复合中心。近年来,研究者们对α-Fe2O3表面进行多种修饰来加速表面反应动力学,提高电子空穴的分离效率,这些合理的方法都对α-Fe2O3的光电催化性能产生了积极的影响。
4.2.1 负载助催化剂
应用电沉积的方法将Co-Pi复合物负载到α-Fe2O3的表面可有效降低析氧反应过电势,原因在于Co-Pi促进光生空穴从α-Fe2O3向界面处的转移,并钝化了表面缺陷,抑制了电子-空穴在界面处的复合[58-59],如图9。
TILLEY等[60]用IrO2纳米颗粒胶体电沉积至α-Fe2O3表面,提高了材料表面的析氧反应动力学,在AM 1.5G的光照强度下,外加电压1.23V(RHE)时,光电流密度超过了3mA/cm2,主要原因是修饰后的材料表面有一个过渡带,正是这个过渡带提供能量促进了空穴的转移。铱的金属有机物也可电沉积到α-Fe2O3表面,形成一层IrOx薄膜[61],随着薄膜中Ir含量的增加,过电势逐渐降低,并且比IrO2纳米颗粒修饰后的过电势更低。相对于IrOx贵金属氧化物,廉价的NiFeOx也能起到相似的效果,负载到表面这层约100nm厚的非晶态覆盖层可以降低固-液界面处的亥姆霍兹层产生的过电势,改变材料表面费米能级的位置,增加了光照产生的电压,促进了析氧反应的热力学[62]。另外,两种廉价的金属锌和钴是以复合氢氧化物的形式被电沉积到α-Fe2O3表面,这种复合双金属氢氧化物以片状的形式垂直生长在催化剂表面,增加了电极与电解液的接触面积,减少了电子-空穴在界面处的复合,它本身具有优良的阳极电催化性能,可以加速空穴氧化水的动力学[63]。

图9 用90mC/cm2的电荷量电沉积得到的Co-Pi薄膜扫描电镜图[59]
借助原子气相沉积技术,可在赤铁矿表面沉积一层极薄透明的Co(OH)2/Co3O4混合物[64],厚度仅相当于电沉积Co-Pi的千分之一,与经过IrO2纳米颗粒修饰后的材料相比,稳定性更加突出;经过电化学阻抗(EIS)分析,这层薄膜可降低固-液界面的接触电阻,加快电荷转移的动力学,又由于厚度较薄,难以起到表面钝化的作用。在水热合成α-Fe2O3的前体时,加入硝酸钴可以实现对α-Fe2O3表面的原位修饰,这种原位修饰的方法使表面沉积的Co3O4颗粒很小,具有较高的粗糙度[65]。WENDER等[66]在纳米环状的α-Fe2O3表面负载一层Co(OH)2,无需外加偏压即可实现对水的分解,产氢效率比TiO2高出许多,表面的这层助催化剂有可能改变了α-Fe2O3的导带位置。
在电化学中,羟基氧化铁可被用作阳极析氧催化剂;用水热法可将β-FeOOH以纳米针的形状生长在α-Fe2O3纳米棒表面,纳米针充当了纳米棒的“枝”,这种微观修饰可降低电解液与催化剂在界面处的阻抗,加速电子的转移,同时它也提高了平带电势[67]。用光辅助电沉积的方法,可将电催化活性更高的非晶态FeOOH负载到α-Fe2O3表面[68],这种纳米结构的薄膜具有高的反应面积,可以提高空穴的有效浓度,加速界面处的氧化动力学,降低了开口电压;用相同的方法在α-Fe2O3表面光沉积一层非晶态的NiOOH,其光电催化性能也得到明显改善。
在酸性溶液中,α-Fe2O3表面会被腐蚀,获得类似负载催化剂的效果,降低了开口电压,但是这种表面处理并没有加速氧化水的动力学,只是降低了光催化分解水反应的逆反应速率[69]。
4.2.2 表面钝化
表面钝化处理可有效降低α-Fe2O3表面处的复合中心浓度。α-Ga2O3和α-Fe2O3都是刚玉型晶体结构,晶格的不匹配度极小,当两种材料复合时,作为负载物的α-Ga2O3会释放α-Fe2O3表层的晶格应力,从而降低陷阱的浓度,并且会保持光电流的稳定性[70]。DIAB等[71]用不同的钴源对多孔的α-Fe2O3薄膜进行表面处理,热分解后产生的CoO钝化了催化剂的表面,而且乙酸钴比硝酸钴的修饰效果更好。TiO2作为光催化剂,同样可以钝化α-Fe2O3表面,增加光生电压,使光电流密度提升4倍[13]。以上这些表面钝化处理一般都会提高光电流的稳定性。
4.2.3 表面功能化
在α-Fe2O3表面可以外接一些基团或与其他材料结合形成特殊的结构,这些功能化的处理虽然没有加速氧化反应和降低表面缺陷的浓度,但是它们可以改变催化剂表面的电荷状态或者促进电荷的转移。
在α-Fe2O3表面沉积一层ZnO,热处理时两种物质可以在界面处反应形成铁酸锌[Zn(Fe2O4)][72],但是并没有起到表面钝化的作用,而是与基底形成异质结构,减少电荷在界面处的积聚,促进电荷的分离。运用原子层沉积技术,也可在β-FeOOH的表面沉积厚度为4nm的TiO2,热处理后可在α-Fe2O3的表面形成一层钛酸铁(Fe2TiO5),提高了电子-空穴在此表面结处的分离效率,使得光电流密度提升了3.5倍[73]。
在用氟化物修饰钛掺杂的α-Fe2O3时,氟离子会与催化剂表层中的钛元素形成价键连接在一起[74],如式(10)。
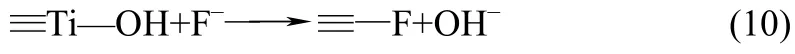
附着在表面的氟离子起到了类似官能团的作用,这种修饰的结果降低了平带电势,使得α-Fe2O3的导带位置低于析氢电势,无需外加偏压,就能分解水。
磷酸根修饰后的α-Fe2O3可以形成负的静电区域,在这层区域中,光生电子-空穴可以有效地被分离,并且促进空穴向电解液转移。这种修饰总的结果是提高了电流密度,但是没有降低开口电势,说明表面的磷酸根并没有起到助催化剂的作用[75]。
NEUFELD等[76]用密度泛函理论模拟了在α-Fe2O3表面覆盖一层α-Al2O3的结果,在部分覆盖时,光生空穴被局域在表面的氧原子附近,而α-Al2O3相对于α-Fe2O3有较高的价带边缘,可以促进空穴的转移,降低外加电压。
可以应用多种方法和物质对α-Fe2O3进行表面处理,但是基本可以分为以下三类:一是负载析氧催化剂,这种方法可以提高阳极反应的动力学,降低开口电压,提高光电流密度,是一种行之有效的方法;二是表面钝化,它可以降低表面缺陷的浓度,降低电子-空穴复合的概率,提高光电流的稳定性;三是对表面类似功能化的处理,这类修饰相对少见,但是被认为是最有发展前景的方法之一[76]。
4.3 复合材料
除了元素掺杂和表面处理外,α-Fe2O3也可与其他材料结合形成复合材料,引入的这些材料可以调控α-Fe2O3的生成形貌,扩大光吸收的表面积,增加光生电压,提高光能-电流转化效率等。
4.3.1 设置衬层
硅的氧化物(SiOx)可以作为α-Fe2O3的衬层沉积到FTO上,这层极薄的衬层可使沉积其上的α-Fe2O3具有精细的纳米结构,厚度仅为12.5nm[77]。以Nb2O5为衬层,可以得到厚度更薄、性能更佳的α-Fe2O3;与SiOx不同,这层厚度仅为2nm的Nb2O5难以影响到α-Fe2O3的形貌,但是它活化了表面的α-Fe2O3,并且α-Fe2O3导带的边缘要比Nb2O5的高约0.2eV[78],这为电子单向流向Nb2O5提供了额外的动力,有利于提高电子-空穴的分离效率。
α-Fe2O3也可与贵金属Au纳米孔[79]、纳米棒[80]复合。在和金纳米孔复合时,α-Fe2O3纳米棒长在金纳米孔中,类似于微型的光学纤维,增强子和等离子体的能量转移,拓宽了对光谱的吸收范围,而且长程有序的金纳米孔在与α-Fe2O3的界面处会产生强烈的局域等离子效应,延长了电荷载体的寿命,使得光电流密度增加了10倍。刘东等[81]将20nm厚的α-Fe2O3与超薄的银纳米孔阵列组成双层结构,极大地减小了光生电子-空穴的复合概率,同时亚波长共振效应和局部表面等离子激元效应强化了对可见光的吸收,使得在相同厚度下α-Fe2O3的光电流密度提升了两倍多。
4.3.2 增大表面积
单相的α-Fe2O3导电性能较差,如果仅以自身形貌的调节来增加光吸收的面积,难以明显地提高光电催化性能,所以可以将α-Fe2O3与导电性能良好、表面积大的材料复合。石墨烯具有二维的网状结构,几乎透明,导电率较高,α-Fe2O3经常与其形成复合材料[82];这种复合材料一般具有较大的表面积,可以促进光能的吸收和增加与电解液的接触面积,在外加偏压的作用下,其中的石墨烯能收集光生电子,延缓电子-空穴的复合,因此,这种复合材料可以显著地增加光电流密度。LIN等[18]运用原子层沉积技术,在TiSi2合金纳米网上沉积α-Fe2O3,这层纳米网起到了结构支撑和电荷收集的作用,弥补了α-Fe2O3的不足。YANG等[83]用化学气相沉积法把α-Fe2O3沉积到ITO纳米棒表面,构建了核-壳结构,它比单一的α-Fe2O3纳米棒对光谱的吸收作用更强,空穴的收集效率更高,也明显地提高了催化剂的稳定性。在三维纳米光子结构[84]的表面沉积一层85nm厚α-Fe2O3,得到复合材料的光电流密度为3.05mA/cm2,大约为平板电极的3倍,表现出了优异的光电催化性能,原因是这种三维结构是由多种材料(Al-Al2O3-Ti/Pt-FTO)组合形成的纳米针阵列,纳米针的长度可以达到1000nm,促进了表面α-Fe2O3对光谱的吸收,如图10。
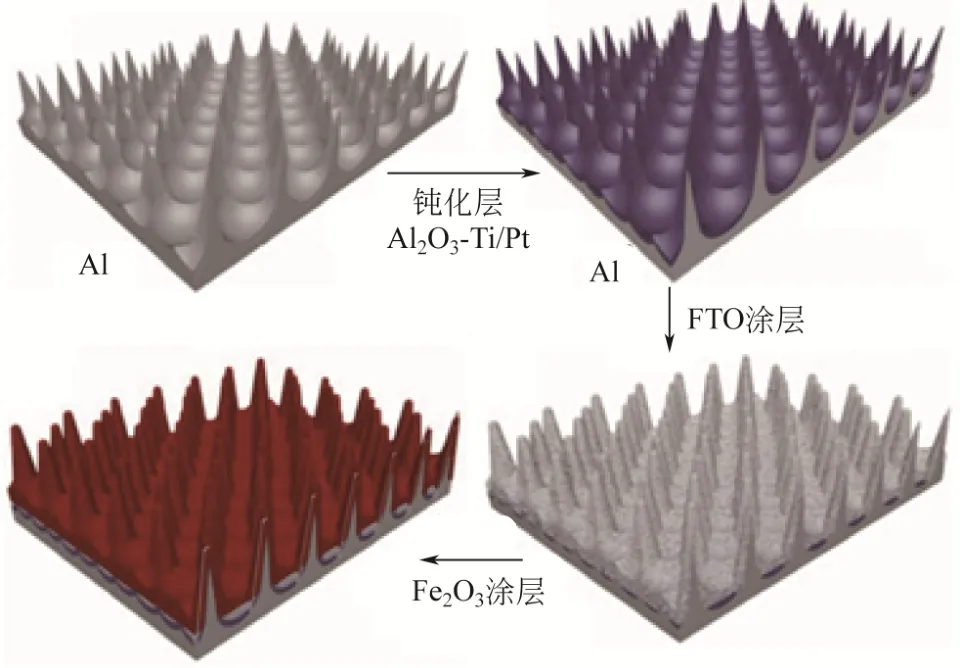
图10 三维纳米针阵列电极示意图[84]
4.3.3 提高光生电压
为提高对太阳能的利用率,增加光生电压,α-Fe2O3经常与其他光催化材料复合。TiO2是目前人们研究最多的材料之一,可以与α-Fe2O3复合,但是TiO2的带隙横跨了α-Fe2O3的带隙,造成了价带的不匹配,使得光生电子-空穴易复合。LIN等[85]提高了核-壳结构中核TiO2价带的位置,缩短了与α-Fe2O3的带隙差距,并在壳(α-Fe2O3)的表面制造了丰富的氧空缺活性位点和OH-桥键,使得光生空穴能够定向移动。ZnO与TiO2带隙相近,同样存在能带不匹配的问题。相对于TiO2和ZnO宽带隙的光催化剂,WO3(2.6eV)与α-Fe2O3复合[86],可以延长光生空穴的寿命,降低开口电压。硅是优良的半导体,带隙宽度为1.1eV,能够吸收600~1200nm波长的光,与α-Fe2O3组成复合材料,可以拓宽对太阳能光谱的吸收范围,使开口电压低至0.6V(RHE);复合材料中的界面成为α-Fe2O3光生电子和Si纳米线光生空穴的复合中心,保证了电流在复合材料中的单向流动,实现了两种材料的有效结合。QI等[87]用n-Si与α-Fe2O3复合构筑了核-壳异质结构,使开口电压进一步降低至0.5V;在此异质结构中,壳(α-Fe2O3)抬高了核(n-Si)的能带边缘能级,同时核也提高了壳的光生电荷分离效率,如图11。
在与α-Fe2O3复合的材料中,可以主要分为两类:一是作为衬层或结构支撑,这类物质不能产生光响应,但是可以调节表面α-Fe2O3的形貌或收集α-Fe2O3的光生电荷;二是本身作为光催化剂与α-Fe2O3复合,增加了光生电压,提高了对光能的利用率。
5 总结与展望
α-Fe2O3廉价、无毒性、稳定性好、具有合适的带隙,理论光电分解水的效率较高,被认为是最具有发展前景的材料之一。但是,它的导电性差、电荷载体扩散长度短、析氧反应速率慢阻碍了其发展。元素掺杂、表面处理等都是改善其不足,提高光电流密度的有效方法,而且这些方法之间存在着一定的关系:元素的掺杂不仅能够提高α-Fe2O3的导电性,还会影响形貌的生成(如Si);掺杂元素还容易在半导体的表面出现富集的状况,影响到表面的催化氧化过程;纳米结构增大了催化剂与电解液的接触面积,提高反应速率。与其他材料复合是提高α-Fe2O3光生电荷的收集效率,拓宽对太阳能光谱的吸收范围,减小外加电压的重要方法。
经过二十年左右的不断发展,α-Fe2O3的光电流密度不断地得到提高,但是单一方法的应用对α-Fe2O3性能的提升有限,未来对材料的改性应该是多种手段的有机结合。KIM等[88]用Pt和Co-Pi分别对α-Fe2O3进行掺杂和表面修饰,使光电流密度提高到新的水平(4.32mA/cm2)。在与n-Si纳米线复合时,光生电流密度更是超过了5mA/cm2,如图12。相信在不久的将来,随着多种改性方法的有效综合应用,α-Fe2O3的光电催化性能会继续得到提升,进一步迈向应用。

图11 光照条件下,n-Si/α-Fe2O3在电解液中电荷转移的能带示意图[87]

图12 在标准条件下(AM1.5G和1.23Vvs. RHE),α-Fe2O3作为光阳极催化分解水电流密度提高的过程[88]
[1] PRVOT M S,SIVULA K. Photoelectrochemical tandem cells for solar water splitting[J]. J. Phys. Chem. C,2013,117(35):17879-17893.
[2] IORDANOVA N,DUPUIS M,ROSSO K M. Charge transport in metal oxides:a theoretical study of hematite α-Fe2O3[J]. The Journal of Chemical Physics,2005,122(14):144305.
[3] HARDEE K L,BARD A J. Semiconductor electrodes:Ⅴ. The application of chemically vapor deposited iron oxide films to photosensitized electrolysis[J]. J. Electrochem. Soc.,1976,123(7):1024-1026.
[4] CARVALHO V,LUZ R A D S,LIMA B H,et al. Highly oriented hematite nanorods arrays for photoelectrochemical water splitting[J]. J. Power Sources,2012,205:525-529.
[5] YEH L S R,HACKERMAN N. Iron oxide semiconductor electrodes in photoassisted electrolysis of water[J]. J. Electrochem. Soc.,1977,124(6):833-836.
[6] FORMAL F L,T TREAULT N,CORNUZ M,et al. Passivating surface states on water splitting hematite photoanodes with alumina overlayers[J]. Chem. Sci.,2011,2(4):737-743.
[7] IANDOLO B,WICKMAN B,ZORIĆ I,et al. The rise of hematite:origin and strategies to reduce the high onset potential for the oxygen evolution reaction[J]. J. Mater. Chem. A,2015,3(33):16896-16912.
[8] MA Y,JOHNSON P D,WASSDAHL N,et al. Electronic structures of α-Fe2O3and Fe3O4from ok-edge absorption and emission spectroscopy[J]. Phys. Rev. B,1993,48(4):2109-2111.
[9] BOSMAN A J,VAN DAAL H J. Small-polaron versus band conduction in some transition-metal oxides[J]. Adv. Phys.,2006,19(77):11-17.
[10] KUDO A,MISEKI Y. Heterogeneous photocatalyst materials for water splitting[J]. Chem. Soc. Rev.,2009,38(1):253-278.
[11] BEERMANN N,VAYSSIERES L,LINDQUIST S-E,et al. Photoelectrochemical studies of oriented nanorod thin films of hematite[J]. J. Electrochem. Soc.,2000,147(7):2456-2461.
[12] PHUAN Y W,CHONG M N,ZHU T,et al. Effects of annealing temperature on the physicochemical,optical and photoelectrochemical properties of nanostructured hematite thin films preparedviaelectrodeposition method[J]. Mater. Res. Bull.,2015,69:71-77.
[13] AHMED M G,KRETSCHMER I E,KANDIEL T A,et al. A facile surface passivation of hematite photoanodes with TiO2overlayers for efficient solar water splitting[J]. ACS Appl. Mater. Interfaces,2015,7(43):24053-24062.
[14] CARVALHO-JR W M,SOUZA F L. Thermal enhancement of water affinity on the surface of undoped hematite photoelectrodes[J]. Sol. Energ. Mat. Sol. C.,2016,144:395-404.
[15] KAY A,CESAR I,GR TZEL M. New benchmark for water photooxidation by nanostructured α-Fe2O3films[J]. J. Am. Chem. Soc.,2006,128(49):15714-15721.
[16] WARREN S C,VO TCHOVSKY K,DOTAN H,et al. Identifying champion nanostructures for solar water-splitting[J]. Nat. Mater.,2013,12(9):842-849.
[17] KLAHR B M,MARTINSON A B,HAMANN T W. Photoelectrochemical investigation of ultrathin film iron oxide solar cells prepared by atomic layer deposition[J]. Langmuir,2011,27(1):461-468.
[18] LIN Y,ZHOU S,SHEEHAN S W,et al. Nanonet-based hematite heteronanostructures for efficient solar water splitting[J]. J. Am. Chem. Soc.,2011,133(8):2398-401.
[19] WANG T,LUO Z,LI C,et al. Controllable fabrication of nanostructured materials for photoelectrochemical water splittingviaatomic layer deposition[J]. Chem. Soc. Rev.,2014,43(22):7469-7484.
[20] SARTORETTI C J,ALEXANDER B D,SOLARSKA R,et al. Photoelectrochemical oxidation of water at transparent ferric oxide film electrodes[J]. Physical Chemistry B,2005,109(28):13685-13692.
[21] SCHREBLER R,LLEWELYN C,VERA F,et al. An electrochemical deposition route for obtaining α-Fe2O3thin films[J]. Electrochemical and Solid-State Letters,2007,10(10):D95.
[22] SPRAY R L,CHOI K S. Photoactivity of transparent nanocrystalline Fe2O3electrodes preparedviaanodic electrodeposition[J]. Chem. Mater.,2009,21(15):3701-3709.
[23] SHINDE P S,GO G H,LEE W J. Facile growth of hierarchical hematite (α-Fe2O3) nanopetals on FTO by pulse reverse electrodeposition for photoelectrochemical water splitting[J]. J. Mater. Chem.,2012,22(21):10469.
[24] MAO A,SHIN K,KIM J K,et al. Controlled synthesis of vertically aligned hematite on conducting substrate for photoelectrochemical cells:nanorods versus nanotubes[J]. ACS Applied Materials & Interfaces,2011,3(6):1852-1858.
[25] PRAKASAM H E,VARGHESE O,PAULOSE M,et al. Synthesis and photoelectrochemical properties of nanoporous iron (Ⅲ) oxide by potentiostatic anodization[J]. Nanotechnology,2006,17(17):4285.
[26] FAN Z,WEN X,YANG S,et al. Controlled p- and n-type doping of Fe2O3nanobelt field effect transistors[J]. Appl. Phys. Lett.,2005,87(1):13113.
[27] SIVULA K,ZBORIL R,FORMAL F L,et al. Photoelectrochemical water splitting with mesoporous hematite prepared by a solution-based colloidal approach[J]. J. Am. Chem. Soc.,2010,132(21):7436-7444.
[28] JIA C J,SUN L D,YAN Z G,et al. Single-crystalline iron oxide nanotubes[J]. Angewandte Chemie,2005,117(28):4402-4407.
[29] WU Z,YU K,ZHANG S,et al. Hematite hollow spheres with a mesoporous shell:controlled synthesis and applications in gas sensor and lithium ion batteries[J]. J. Phys. Chem. C,2008,112(30):11307-11313.
[30] ZENG S,TANG K,LI T,et al. Facile route for the fabrication of porous hematite nanoflowers:its synthesis,growth mechanism,application in the lithium ion battery,and magnetic and photocatalytic properties[J]. J. Phys. Chem. C,2008,112(13):4836-4843.
[31] PENDLEBURY S R,BARROSO M,COWAN A J,et al. Dynamics of photogenerated holes in nanocrystalline α-Fe2O3electrodes for water oxidation probed by transient absorption spectroscopy[J]. Chem. Commun.,2011,47(2):716-718.
[32] SEGEV G,DOTAN H,MALVIYA K D,et al. High solar flux concentration water splitting with hematite (α-Fe2O3) photoanodes[J]. Advanced Energy Materials,2016,6(1):DOI:10.1002/aenm. 201500817.
[33] LIN Y,XU Y,MAYER M T,et al. Growth of p-type hematite by atomic layer deposition and its utilization for improved solar water splitting[J]. J. Am. Chem. Soc.,2012,134(12):5508-5511.
[34] GLASSCOCK J A,BARNES P R F,PLUMB I C,et al. Structural,optical and electrical properties of undoped polycrystalline hematite thin films produced using filtered arc deposition[J]. Thin Solid Films,2008,516(8):1716-1724.
[35] CESAR I,SIVULA K,KAY A,et al. Influence of feature size,film thickness,and silicon doping on the performance of nanostructured hematite photoanodes for solar water splitting[J]. J. Phys. Chem. C,2009,113(2):772-782.
[36] KUMARA P,SHARMAA P,SHRIVASTAV R,et al. Electrodeposited zirconium-doped α-Fe2O3thin film for photoelectrochemical water splitting[J]. Int. J. Hydrogen. Energ.,2011,36(4):2777-2784.
[37] SHEN S,KRONAWITTER C X,WHEELER D A,et al. Physical and photoelectrochemical characterization of Ti-doped hematitephotoanodes prepared by solution growth[J]. J. Mater. Chem. A,2013,1(46):14498-14506.
[38] GURUDAYA L,CHIAM S Y,KUMAR M H,et al. Improving the efficiency of hematite nanorods for photoelectrochemical water splitting by doping with manganese[J]. ACS Appl. Mater. Interfaces,2014,6(8):5852-5859.
[39] LIU J,CAI Y Y,TIAN Z F,et al. Highly oriented Ge-doped hematite nanosheet arrays for photoelectrochemical water oxidation[J]. Nano Energy,2014,9:282-290.
[40] HAHN N T,YE H,FLAHERTY D W,et al. Reactive ballistic deposition of α-Fe2O3thin films for photoelectrochemical water oxidation[J]. ACS Nano,2010,4(4):1977-1986.
[41] REICHMAN J. The current-voltage characteristics of semiconductorelectrolyte junction photovoltaic cells[J]. Appl. Phys. Lett.,1980,36(7):574-577.
[42] KLAHR B,GIMENEZ S,FABREGAT-SANTIAGO F,et al. Water oxidation at hematite photoelectrodes:the role of surface states[J]. J. Am. Chem. Soc.,2012,134(9):4294-302.
[43] GURLO A,BÂRSAN N,OPREA A,et al. An n- to p-type conductivity transition induced by oxygen adsorption on alpha-Fe2O3[J]. Applied Physics Letters,2004,85(12):DOI:org/10.1063/1.1794853.
[44] LING Y,WANG G,WHEELER D A,et al. Sn-doped hematite nanostructures for photoelectrochemical water splitting[J]. Nano Letters,2011,11(5):2119-2125.
[45] WANG L,LEE C Y,MAZARE A,et al. Enhancing the water splitting efficiency of Sn-doped hematite nanoflakes by flame annealing[J]. Chemistry—A:European Journal,2013,20(1):72-82.
[46] WANG J J,HU Y,TOTH R,et al. A facile nonpolar organic solution process of a nanostructured hematite photoanode with high efficiency and stability for water splitting[J]. J. Mater. Chem. A,2016,
[47] KLEIMAN-SHWARSCTEIN A,HU Y-S,FORMAN A J,et al. Electrodeposition of α-Fe2O3doped with Mo Cr as photoanodes for photocatalytic water splitting[J]. The Journal of Physical Chemistry C,2008,112(40):15900-15907.
[48] BAKA A,CHOIB W,PARK H. Enhancing the photoelectrochemical performance of hematite (α-Fe2O3) electrodes by cadmium incorporation[J]. Applied Catalysis B:Environmental,2011,110:207-215.
[49] LIU Y,YU Y X,ZHANG W D. Photoelectrochemical properties of Ni-doped Fe2O3thin films prepared by electrodeposition[J]. Electrochim. Acta,2012,59:121-127.
[50] ZHANG P,KLEIMAN-SHWARSCTEIN A,HU Y S,et al. Oriented Ti doped hematite thin film as active photoanodes synthesized by facile APCVD[J]. Energy Environ. Sci.,2011,4(3):1020.
[51] FRANKING R,LI L,LUKOWSKI M A,et al. Facile post-growth doping of nanostructured hematite photoanodes for enhanced photoelectrochemical water oxidation[J]. Energy Environ. Sci.,2013,6(2):500-512.
[52] MAO A,PARK N G,HAN G Y,et al. Controlled growth of vertically oriented hematite/Pt composite nanorod arrays:use for photoelectrochemical water splitting[J]. Nanotechnology,2011,22(17):175703.
[53] 洪孝挺,王正鹏,陆峰,等. 可见光响应型非金属掺杂TiO2的研究进展[J]. 化工进展,2004,23(10):1077-108. HONG X T,WANG Z P,LU F,et al. Study on visble light-activated non-metal doped titanium dioxide[J]. Chemical Industry and Engineering Progress,2004,23(10):1077-1080.
[54] 谢先法,吴平霄,党志,等. 过渡金属离子掺杂改性TiO2研究进展[J]. 化工进展,2005,24(12):1358-1362. XIE X F,WU P X,DANG Z,et al. Research progress of photocatalytic performance of TiO2modified by doped transition metal ions[J]. Chemical Industry and Engineering Progress,2005,24(12):1358-1362.
[55] KLEIMAN-SHWARSCTEIN A,HUDA M N,WALSH A,et al. Electrodeposited aluminum-doped α-Fe2O3photoelectrodes:Experiment and theory[J]. Chem. Mater.,2010,22(2):510-517.
[56] LIAO P,KEITH J A,CARTER E A. Water oxidation on pure and doped hematite(0001)surfaces:prediction of Co and Ni as effective dopants for electrocatalysis[J]. J. Am. Chem. Soc.,2012,134(32):13296-309.
[57] PETER L M. Energetics and kinetics of light-driven oxygen evolution at semiconductor electrodes:the example of hematite[J]. J. Solid State Electr.,2012,17(2):315-326.
[58] ZHONG D K,GAMELIN D R. Photoelectrochemical water oxidation by cobalt catalyst (“Co-Pi”)/α-Fe2O3composite photoanodes:oxygen evolution and resolution of a kinetic bottleneck[J]. J. Am. Chem. Soc.,2010,132(12):4202-4207.
[59] KLAHR B,GIMENEZ S,FABREGAT-SANTIAGO F,et al. Photoelectrochemical and impedance spectroscopic investigation of water oxidation with “Co–Pi”-coated hematite electrodes[J]. J. Am. Chem. Soc.,2012,134(40):16693-16700.
[60] TILLEY S D,CORNUZ M,SIVULA K,et al. Light-induced water splitting with hematite:improved nanostructure and iridium oxide catalysis[J]. Angew. Chem. Int. Edit.,2010,49(36):6405-6408.
[61] BADIA-BOU L,MAS-MARZA E,RODENAS P,et al. Water oxidation at hematite photoelectrodes with an iridium-based catalyst[J]. J. Phys. Chem. C,2013,117(8):3826-3833.
[62] DU C,YANG X,MAYER M T,et al. Hematite-based water splitting with low turn-on voltages[J]. Angew. Chem. Int. Edit.,2013,52(48):12692-12695.
[63] XU D,RUI Y,LI Y,et al. Zn-Co layered double hydroxide modified hematite photoanode for enhanced photoelectrochemical water splitting[J]. Appl. Surf. Sci.,2015,358:436-442.
[64] RIHA S C,KLAHR B M,TYO E C,et al. Atomic layer deposition of a submonolayer catalyst for the enhanced photoelectrochemical performance of water oxidation with hematite[J]. ACS Nano,2013,7(3):2396-2405.
[65] XI L,TRAN P D,CHIAM S Y,et al. Co3O4-decorated hematite nanorods as an effective photoanode for solar water oxidation[J]. J. Phys. Chem. C,2012,116(26):13884-13889.
[66] WENDER H,GON ALVES R V,DIAS C S B,et al. Photocatalytic hydrogen production of Co(OH)2nanoparticle-coated α-Fe2O3nanorings[J]. Nanoscale,2013,5(19):9310-9316.
[67] YANG T Y,KANG H Y,JIN K,et al. Iron oxide photoanode with hierarchical nanostructure for efficient water oxidation[J]. J. Mater. Chem. A,2013,2(7):2297-2305.
[68] YU Q,MENG X,WANG T,et al. Hematite films decorated with nanostructured ferric oxyhydroxide as photoanodes for efficient and stable photoelectrochemical water splitting[J]. Adv. Funct. Mater.,2015,25(18):2686-2692.
[69] CAO D,LUO W,FENG J,et al. Cathodic shift of onset potential for water oxidation on a Ti4+doped Fe2O3photoanode by suppressing theback reaction[J]. Energy Environ. Sci.,2014,7(2):752-759.
[70] HISATOMI T,FORMAL F L,CORNUZ M,et al. Cathodic shift in onset potential of solar oxygen evolution on hematite by 13-group oxide overlayers[J]. Energy Environ. Sci.,2011,4(7):2512.
[71] DIAB M,MOKARI T. Thermal decomposition approach for the formation of alpha-Fe2O3mesoporous photoanodes and an α-Fe2O3/CoO hybrid structure for enhanced water oxidation[J]. Inorg. Chem.,2014,53(4):2304-2309.
[72] LUO Z B,LI C C,ZHANG D,et al. Highly-oriented Fe2O3/ZnFe2O4nanocolumnar heterojunction with improved charge separation for photoelectrochemical water oxidation[J]. Chem. Commun.,2016,52(58):9013-9015.
[73] LI C C,WANG T,LUO Z B,et al. Enhanced charge separation through ALD-modified Fe2O3/Fe2TiO5nanorod heterojunction for photoelectrochemical water oxidation[J]. Small,2016,12(25):3415-3422.
[74] HU Y S,KLEIMAN-SHWARSCTEIN A,STUCKY G D,et al. Improved photoelectrochemical performance of Ti-doped α-Fe2O3thin films by surface modification with fluoride[J]. Chem. Commun.,2009(19):2652-2654.
[75] KIM J Y,JANG J W,YOUN D H,et al. A stable and efficient hematite photoanode in a neutral electrolyte for solar water splitting:towards stability engineering[J]. Advanced Energy Materials,2014,4(13):DOI:10.1002/aenm.201400476.
[76] NEUFELD O,YATOM N,CASPARY TOROKER M. A first-principles study on the role of an Al2O3overlayer on Fe2O3for water splitting[J]. ACS Catalysis,2015,5(12):7237-7243.
[77] FORMAL B F L,GR TZEL M,SIVULA K. Controlling photoactivity in ultrathin hematite films for solar water-splitting[J]. Adv. Funct. Mater.,2010,20(7):1099-1107.
[78] HISATOMI T,DOTAN H,STEFIK M,et al. Enhancement in the performance of ultrathin hematite photoanode for water splitting by an oxide underlayer[J]. Adv. Mater.,2012,24(20):2699-2702.
[79] LI J T,CUSHING S K,ZHENG P,et al. Plasmon-induced photonic and energy-transfer enhancement of solar water splitting by a hematite nanorod array[J]. Nat. Commun.,2013,4:DOI:10.1038/ncomms3651.
[80] JUE WANG,PAN S,CHEN M,et al. Gold nanorod-enhanced light absorption and photoelectrochemical performance of α-Fe2O3thin-film electrode for solar water splitting[J]. J. Phys. Chem. C,2013,117(42):22060-22068.
[81] 刘东,于海童,杨震,等. 超薄膜吸收器在太阳能光解水制氢中的应用[J]. 化工学报,2015,66(s1):297-301. LIU D,YU H T,YANG Z,et al. Ultrathin film absorber for solar water splitting[J]. CIESC Journal,2015,66(s1):297-301.
[82] ZHANG K,SHI X,KIM J K,et al. Inverse opal structured α-Fe2O3on graphene thin films:enhanced photo-assisted water splitting[J]. Nanoscale,2013,5(5):1939-1944.
[83] YANG J,BAO C,YU T,et al. Enhanced performance of photoelectrochemical water splitting with ITO@α-Fe2O3core-shell nanowire array as photoanode[J]. ACS Appl. Mater. Interfaces,2015,48(7):26482-26490.
[84] QIU Y,LEUNG S F,ZHANG Q,et al. Efficient photoelectrochemical water splitting with ultrathin films of hematite on three-dimensional nanophotonic structures[J]. Nano Letters,2014,14(4):2123-2129.
[85] LIN Z,LIU P,YAN J,et al. Matching energy levels between TiO2and α-Fe2O3in a core-shell nanoparticle for visible-light photocatalysis[J]. J. Mater. Chem. A,2015,28(3):14853-14863.
[86] ZHAO P,KRONAWITTER C X,YANG X,et al. WO3-α-Fe2O3composite photoelectrodes with low onset potential for solar water oxidation[J]. Phys. Chem. Chem. Phys.,2014,16(4):1327-1332.
[87] QI X,SHE G,HUANG X,et al. High-performance n-Si/α-Fe2O3core/shell nanowire array photoanode towards photoelectrochemical water splitting[J]. Nanoscale,2013,6(6):3182-3189.
[88] KIM J Y,LEE J S,MAGESH G,et al. Single-crystalline,wormlike hematite photoanodes for efficient solar water splitting[J]. Scientific Reports,2013,3:DOI:10.1038/srepo2681.
Hematite photoanodes for solar water splitting
WANG Kaifang,LIU Guang,GAO Xusheng,HE dongying,LI Jinping
(Research Institute of Fine Chemicals,Taiyuan University of Technology,Taiyuan 030024,Shanxi,China)
Photoelectrochemical cell is able to turn sunlight into stored energy conveniently in the form of hydrogen,and the stable and low-cost photoanode catalyst is crucial in this device. Hematite is considered as one of the most promising photoanode catalysts due to its suitable band gap,high theoretical solar to hydrogen efficiency,chemical stability under illumination and rich storage in earth. However,the poor conductivity,short photo-generated charge carrier lifetime and high turn-on voltage have limited the performance improvement of hematite severely. This review introduces the basic mechanism of photoelectrocatalysis and energy band excitation,then it summarizes the synthesis of nanostructure α-Fe2O3and the improvements on the photoelectrocatalysis property of hematite in recent years,including conductivity enhancement by element doping,oxygen evolution overpotential or trap concentration reduction by surface treatment,and photo-induced voltage or specific area increase by coupling with other materials. The future developing perspectives of hematite are also presented,and multi-modified technologies are considered as important ways to improve the photocurrent density.
hematite;solar energy;photoelectrocatalysis;hydrolysis;hydrogen
O614.81;O644.16;TQ116.2
:A
:1000–6613(2017)02–0397–13
10.16085/j.issn.1000-6613.2017.02.001
2016-05-10;修改稿日期:2016-06-13。
国家自然科学基金(51402205)及山西省基础研究计划(2015021058)项目。
王开放(1990—),男,硕士研究生,研究方向为光电催化。联系人:李晋平。E-mail:jpli211@hotmail.com。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
车用发动机(2021年5期)2021-10-31
石油化工高等学校学报(2021年3期)2021-07-15
防爆电机(2020年4期)2020-12-14
河北理科教学研究(2020年1期)2020-07-24
物理化学学报(2019年8期)2019-09-03
小型内燃机与车辆技术(2016年4期)2016-10-21
车用发动机(2015年2期)2015-04-25
高中生学习·高二版(2014年5期)2014-07-03
