
血清多肽组及其个体差异的纳升液相色谱—高分辨串联质谱分析
2017-02-06 21:37孔祥怡石锴丛乐乐王静蒋立军洪晓
分析化学 2017年1期
孔祥怡+石锴+丛乐乐+王静+蒋立军+洪晓愉+李水明+王勇+赵晴

摘 要 分析和比较疾病组及健康对照组的混合样品是血清多肽组生物标记物研究的常用方法,但对健康个体多肽组的差异和共性关注较少。本研究利用纳升液相色谱.高分辨四级杆飞行时间质谱鉴定健康人混合血清样品(20例)的多肽组,阐明血清多肽组的分子量分布等一般特征,进而选取6例个体样品单独分析并与混合样品的分析结果进行比较,说明正常健康样品之间的个体差异和共同成分。结果表明,可鉴定序列的血清多肽组的分子量范围在7000 Da以下,纤维蛋白原α链等蛋白质所属肽段的检出频率最高,肽段在蛋白质水平上分布具有不均一性,排在前10%的蛋白质占据了约50%的总肽段,而后40%的蛋白质只有1条检出肽段。此外,在所有样品中都检测到了来自于8个蛋白质的12个共同肽段,检测到了N端乙酰化、氨基酸氧化、磷酸化、脱氨化和脱水等翻译后修饰和明显的阶梯序列现象。本研究在肽段序列水平分析了血清多肽组的基本特征和个体差异,可为血清多肽组生物标志物研究提供参考。
关键词 多肽组; 高分辨飞行时间质谱; 血清; 个体差异
1 引 言
生物体内的内源性多肽为蛋白质降解的产物,反映了蛋白质的降解过程,有些多肽还具有抗菌抗炎和信号传导等作用[1]。多肽组学的概念最早出现于2001年,是指生物样品中的全部肽段,生物标记物和神经活性肽研究是多肽组学的两个最主要应用[2]。生物标记物的一个主要来源是血液,随着蛋白质组学技术的完善和拓展,血清多肽组已成为基于质谱的血液生物标记物研究的重要方法。
基质辅助激光解吸电离.飞行时间质谱(MALDI.TOF)和纳升液相色谱.电喷雾串联质谱是进行多肽组研究的两种常用技术,但它们取得的多肽组数据及应用模式不同。MALDI.TOF方法检测的是肽段分子量,在多肽组中也称为谱图轮廓(Spectra profile)[3~5]。利用MALDI.TOF方法筛选生物标记物首先需要在疾病组与对照组中找到相同分子量的质谱峰,然后比较它们离子强度的差异 [6,7],本方法的优势在于简单、直观、质量检测范围较宽,并且MALDI源与傅里叶变换离子回旋共振质谱或串联飞行时间质谱(TOF/TOF)联用也可以获得序列信息[8,9]。但是,在MALDI.TOF/TOF仪器中非酶切肽段的断裂效率通常不够充分,此外,使用MALDI源质谱获得的多肽组的体量相对较小,这可能与MALDI.TOF仪器较少与液相色谱联用和MALDI源中的电离抑制作用有关,例如,即使对于蛋白质标准品的酶切产物也较难检测到全部的酶切肽段[10]。利用纳升液相色谱—串联质谱技术则不仅显著增加了肽段鉴定能力和数目,也可以与标记或非标记技术联用实现相对定量[11,12],但是,尽管该技术拓展了血清多肽组的范围,在应用模式上同样为比较疾病组和健康对照组的混合样品,对个体差异重视不够,目前,尚未有关健康个体样品血清多肽组差异程度的报道。
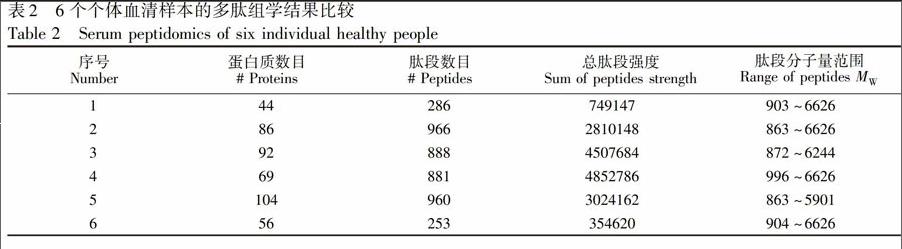
本研究利用氧化石墨烯.磷酸镧纳米磁性复合材料(LaGM)分离和富集血液多肽[13],利用纳升液相色谱.高分辨串联质谱(Nano LC.TripleTOFTM 5600)首先分析健康人的混合血清样本,确定多肽组的分子量范围、肽段分布和磷酸化等翻译后修饰特征,然后对比分析另外6个健康个体的血清样品,探讨血清多肽组的个体差异和共性。
2 实验部分
2.1 仪器与试剂
Eksigent nanoLC.UltraTM 2D 系统、TripleTOF 5600+高分辨质谱仪、Protein Pilot 4.5软件(AB SCIEX); 真空冷冻干燥机(Thermo Savant)。
氧化石墨烯.磷酸镧纳米磁性复合材料,纳升液相色谱流动相 A为0.1% 甲酸.2% 乙腈,流动相 B为0.1% 甲酸.98% 乙腈,C 18反相色谱捕集柱、C 18反相色谱分析柱。
2.2 多肽的分离和富集
30 μL人血清样品溶解于 500 μL去离子水,与20 μL 30 mg/mL LaGM 复合材料混合,在冰水浴中涡旋2 min,1000 r/min振荡10 min。1000 r/min离心5 min,磁分离,在沉淀物中加入500 μL 水,涡旋1 min,振荡5 min,磁分离,去掉上层清液,重复2次。在沉淀中加入20 μL 80%乙腈+1% 三氟乙酸的洗脱液,涡旋1 min,振荡5 min,磁分离,收集上层清液并冷冻干燥。
2.3 反相色谱.TripleTOF质谱分析
将分离冻干的多肽样品溶解于Nano.RPLC Buffer A中,在线Nano.RPLC液相色谱在Eksigent nanoLC.UltraTM 2D系统(AB SCIEX)进行,溶解后的样品以2 μL/min的流速上样到C 18预柱上(100 μm×3 cm, 3 μm, 15 nm),然后保持流速冲洗脱盐10 min。 分析柱是C 18反相色谱柱(75 μm×3 cm,3 μm, 12 nm, ChromXP Eksigent),梯度洗脱条件: 0~42 min,5%~25% B; 42~56 min,25%~40% B; 56~64 min,80% B; 64~70 min,5% B。质谱采用TripleTOF 5600+ 系统(AB SCIEX)。纳升喷雾III离子源(AB SCIEX, USA),喷雾电压为2.4 kV,气帘气压为207 kPa,雾化气压为34.5 kPa,加热温度为150℃,质谱扫描方式为数据依赖的采集工作模式(IDA information dependent analysis),一级TOF.MS单张图谱扫描时间为250 ms,每次IDA循环下最多采集35个电荷为2+~7+且单秒计数大于100的二级图谱,每张二级图谱的累积时间为80 ms。
2.4 数据分析条件
质谱采集到的原始wiff图谱文件,采用Protein pilot software v. 4.5软件进行数据加工处理和检索分析,数据库为Uniprot库中的Homo sapiens人种专一数据库(包含20210条蛋白质序列, 2015年1月2日下载), 检索参数设置为非酶切、磷酸化强调和生物学修饰,数据库: uniprot_Homo sapiens(20210条蛋白质序列),假阳性率控制为1% FDR。
3 结果与讨论
3.1 血清多肽组特征及其在蛋白质水平上的分布
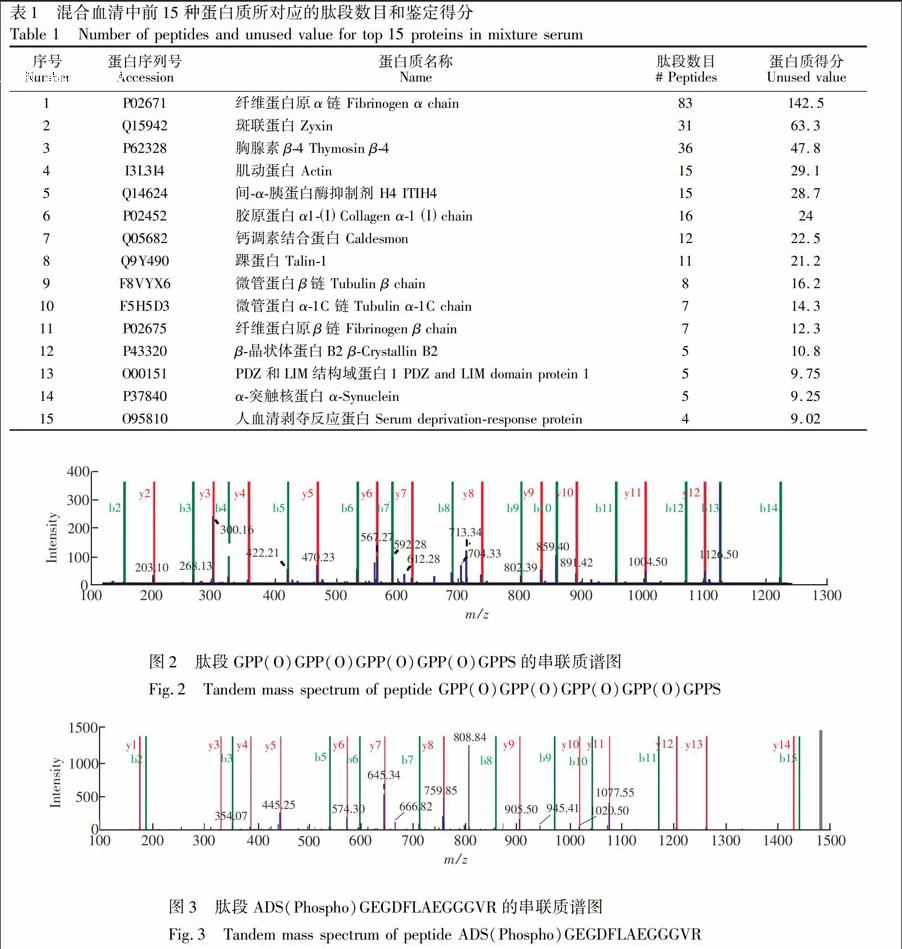
在本研究中将序列相同但翻译后修饰不同的肽段视为不同肽段, 共检测到归属于64个蛋白质的361条特异性肽段, 95%置信度下这些蛋白质覆盖率为从1%变化至97%。它们的质荷比m/z 402~1046, 肽段电荷数为2~7, 等电点3.37~12.01, 一级质量误差均小于5 ppm, 分子量分布为863~6500, 以1~2 kDa的肽段数目为最多, 但在7 kDa分子量以下均有肽段被鉴定(图1)。与文献[14]一致, 本研究也发现肽段在蛋白质水平上并不是平均分布。例如, 搜库得分第一位的纤维蛋白原α链有83条肽段, 是总检出肽段数目的23%, 排位前20%的蛋白质占有了68%以上的总肽段数, 而排名在后40%的26种蛋白质都只有1条肽段被检测。在此混合样品中, 鉴定排名在前15位的蛋白质及其相应的肽段鉴定数目如表1所示。此外, 检出到了磷酸化、侧链氨基酸残基氧化、N端焦谷氨酸化、脱水和脱氨等多种翻译后修饰现象, 例如, 在此样本集中共检测到26个氧化肽段和11个磷酸化肽段, 例如, 肽段GPPGPPGPPGPPGPPS来自于胶原蛋白α.1链, 其氮端3, 6, 9和12位的脯氨酸残基均被氧化, 由于在串联质谱中鉴定到了b2~b14和y2~y12的连续碎片离子(图2), 所以脯氨酸氧化位点可准确给出。肽段ADSGEGDFLAEGGGVR来自于纤维蛋白原α链, 在其N端3位的丝氨酸残基上发生了磷酸化, 同样观察到了近似连续的b和y系列离子(图3), 说明鉴定结果可靠。
3.2 血清多肽组的个体差异
多肽组中发现生物标志物的常用方法是分别分析疾病组和对照组的混合样品,以寻找差异进而进行个体验证,但是,对健康个体样品的分析则较少,本研究以6个样本为例探讨血清多肽组的个体差异和共性。首先比较它们的蛋白质数目、肽段数目、信号强度和分子量范围等指标(表2)。结果表明,这些指标在不同样品之间结果差异很大,例如,鉴定的最少和最多的肽段数目分别为253和966个,而最少和最多的蛋白质数目分别44和104个。值得注意的是,同一血清样品的肽段数目和蛋白质数目并没有严格的对应关系,例如253个肽段对应56个蛋白质,而966个肽段则归属于86个蛋白质。在6例个体样本中,肽段数目与蛋白质数目的比值为4.5~13,这也反映了血清多肽组的个体差异。此外,样品6的信号强度与样品3和4相差一个数量级,而该值相差3倍以上可认为肽段含量有显著性差别[15],由于样品用量、分离方法和仪器条件均为相同设置,本研究认为这些条件的波动不会对结果造成这么大的影响,而是由于血清样品间的多肽组成差异和浓度不同所导致。
3.3血清多肽组的共性
尽管不同样品血清多肽组的结果存在以上差异,但是,也具有一些共同特点。首先,分子量范围接近。多肽组学的质量范围是一个相对宽泛的概念,Shen等[16]认为分子量<3 kDa,使用MALDI.TOF可很容易将分子量范围拓展至10 kDa[17],而若将多肽组理解为蛋白质的降解产物,则其分子量可能会更大。但就序列鉴定而言,尽管很早就有报道Q.TOF类仪器能鉴定分子量8.6 kDa以上的肽段[18],但是通常液质联用技术鉴定的肽段分子量在4000以下[11,12,19],本研究鉴定的所有个体和混合血清样本多肽组的肽段分子量范围都在800~7000之间。TPDVSSALDKLKEFGNTLEDKARELISRIKQSELSAKMREW.FSETFQKVKEKLKIDS为最长肽段,归属于载脂蛋白 C.I,质荷比为1105.4,带6个正电荷,分子量为6632.5; 进一步分析质谱数据,多肽组中还存在分子量更大的肽段,但因断裂不够而未能鉴定序列。其次,在蛋白质水平上有一些蛋白质出现频率很高,在所有个体样品和混合样品中都可以检测到,例如纤维蛋白原α链、胸腺素β.4、斑联蛋白和血清白蛋白等9种蛋白质,归属于这些蛋白质的肽段在序列上可能非常相近,例如,只相差一个氨基酸残,提示血清多肽组的序列存在一定变动,此结果说明相比于分子量,序列信息能够更好地反映多肽组结果之间的联系,推测这是当使用不同分子量作为特征值时,各实验室之间的测定结果重现性不佳的原因之一[2]。尽管如此,如
分子量分别为2758.309和2758.492,如此相近的质量不能被MALDI.TOF分辨,这也说明纳升液相色谱.高分辨串联质谱不仅在肽段的鉴定数目上多于MALDI.TOF方法,还可以根据液相色谱保留时间的不同和良好的串联质谱性能而区分分子量相近的肽段。
4 结 论
本研究基于纳升液相色谱.高分辨串联质谱建立了快速、灵敏和高效的血清多肽组学分析方法,阐明了血清多肽组肽段分布的不均一性和分子量分布范围等多肽组基本特征,发现不同个体样本的血清多肽组即在肽段水平存在共同之处,在蛋白质水平也存在明显差异,提示在利用血清多肽组筛选疾病标志物时需适当考虑个体差异和标志物的稳健性,即肽段序列的差异具有一定偶然性,正常样品的血清多肽组结果同样存在差异,提示并不是所有肽段水平上的差异都与疾病相关。
References
1 Tinoco A D, Saghatelian A. Biochemistry, 2011, 50: 7447-7461
2 Schrader M, Schulz.Knappe P, Fricker L D. Eupa. Open. Proteomcs, 2014, 3: 171-182
3 Zapico.Muiz E, Farré.Viladrich A, Rico.Santana N, González.Sastre F, Mora.Brugués J. Pancreas., 2010 , 39(8): 1293-1298
4 Preianò M, Falcone D, Maggisano G, Montalcini T, Navarra M, Paduano S, Savino R, Terracciano R. Clin. Chim. Acta, 2014, 437(1): 120-128
5 Rico Santana N, Zapico Muiz E, Cocho D, Bravo Y, Delgado Mederos R, Martí.Fbregas J. J. Stroke. Cerebrovasc. Dis., 2014 , 23(2): 235-240
6 Zhang J N, Zhou S N, Zheng H, Zhou Y H, Chen F, Lin J X. Biochem. Biophys. Res. Commun., 2012, 421(4): 844-849
7 Zheng H, Li R X, Zhang J N, Zhou S N, Ma Q W, Zhou Y H, Chen F, Lin J X. Sci. Rep., 2014, 4: 7046
8 Nicolardi S, Velstra B, Mertens B J, Bonsing B,Mesker W E. Translational Proteomics, 2014, 2(1): 39-51
9 Hayakawa E, Landuyt B, Baggerman G, Cuyvers R, Lavigne R, Luyten W, Schoofs L. Peptides, 2013, 42: 63-69
10 WANG Yong, LI Shui.Ming, HE Man.Wen. Chinese J. Anal. Chem., 2013, 41(4): 494-499
王 勇, 李水明, 何曼文. 分析化学, 2013, 41(4): 494-499
11 Caseiro A, Ferreira R, Padro A, Quintaneiro C, Pereira A, Marinheiro R, Vitorino R, Amado F. J. Proteome Res., 2013, 12(4): 1700-1709
12 Paolo N, Fredrik L, Giulia R, Alessandra C, Peter J, Roda A. J. Chromatogr. B, 2009, 877: 3127-3136
13 Cheng G, Wang Z G, Liu Y L, Zhang J L, Sun D H, Ni J Z. Chem. Commun., 2012, 48(82): 10240-10242
14 Zheng X, Baker H, Hancock W S. J. Chromatogr. A, 2006, 1120 (1.2): 173-184
15 HONG Xiao.Yu, WANG Hao, XU Jin.Ling, LI Shui.Ming, WANG Yong. Chinese. J. Anal. Chem., 2016, 44(3): 403-408
洪晓愉, 王 浩, 徐金玲, 李水明, 王 勇. 分析化学, 2016, 44(3): 403-408
16 Shen Y F, Liu T, Tolic' N, Petritis B O, Zhao R, Moore R J, Purvine S O, Camp D G, Smith R D. J. Proteome Res., 2010, 9(5): 2339-2346
17 Liu Y, Wei F, Wang F, Li C, Meng G, Duan H, Ma Q, Zhang W. Biochem. Biophys. Res. Commun, 2015, 465(3): 476-480
18 Mhring T, Kellmann M, Jürgens M, Schrader M. J. Mass Spectro., 2005, 40(2): 214-226
19 ZHU Jun, WANG Fang.Jun, DONG Xiao.Li, YE Ming.Liang, ZOU Han.Fa. Scientia Sinica Chimica, 2010, 40(5): 546-555
朱 俊, 王方军, 董小莉, 叶明亮, 邹汉法. 中国科学: 化学, 2010, 40(5): 546-555
Abstract Peptidomic has become a common method of screening biomarkers, but most researches focused on the mixture samples by analyzing and comparing the results of samples from patients and healthy controls. Unfortunately, there is little study about the individual differences among healthy person and the common features of them. Here, to obtain the general characteristics of serum peptidomics including molecular weight distribution, nanoliter liquid chromatography.high resolution tandem mass spectrometry was used to analyze the mixture samples of 20 healthy human serums. Next, six cases of individual samples were analyzed and compared, indicating that there were obvious individual difference and some common features among different samples. We found that the peptides within 7000 Da could be identified and the peptides from fibrinogen α.chain were detected with the highest frequency. Additionally, the distribution of serum peptidome at protein level was heterogeneous; namely, the top 10% protein accounted about 50% of the total peptides and only one peptide was detected for the last 40% proteins. In addition, 12 common peptides arising from 8 proteins were detected in all of the samples. Furthermore, the post.translational modification including N.terminal acetylation, oxidation, phosphorylation, deamination and dehydration, as well as the obvious sequence ladder phenomenon, were detected in all samples. In conclusion, the basic characteristics of peptidomics at the sequence level were explored and the individual difference of serum peptidome was proposed, which could provide a reference for the study of serum peptide biomarkers.
Keywords Peptidome; High resolution time.of.flight mass spectrometry; Serum; Individual differences
猜你喜欢
中国动物保健(2022年2期)2022-05-05
中国现代医生(2022年6期)2022-04-23
中国典型病例大全(2022年9期)2022-04-19
中国药学药品知识仓库(2022年1期)2022-03-23
小学教学参考(综合)(2016年11期)2016-11-14
科技视界(2016年21期)2016-10-17
科技视界(2016年20期)2016-09-29
中国民族民间医药·下半月(2011年4期)2011-09-27
大众医学(2000年1期)2000-06-07
