
水与MOR分子筛复合环境对布洛芬分子手性转变反应共催化的理论研究
2017-01-16 08:40孙永清王佐成汤得怀
复旦学报(自然科学版) 2016年6期
孙永清,王佐成,高 峰,汤得怀
(1. 白城技师学院 基础部,白城 137000; 2. 白城师范学院 物理与电子信息学院,白城 137000;3. 白城师范学院 计算机科学学院,白城 137000)
水与MOR分子筛复合环境对布洛芬分子手性转变反应共催化的理论研究
孙永清1,王佐成2,高 峰2,汤得怀3
(1. 白城技师学院 基础部,白城 137000; 2. 白城师范学院 物理与电子信息学院,白城 137000;3. 白城师范学院 计算机科学学院,白城 137000)
采用量子力学与分子力学组合的ONIOM方法,研究布洛芬限域在水与MOR分子筛复合环境的手性转变.结构研究表明: 1,2个和3个水分子助氢迁移反应的过渡态分子氢键键角不断增大,3个水分子助氢迁移反应的10元环过渡态结构明显偏离平面.反应通道研究发现: 标题反应有a1,a2和b三个通道.a1和a2是经过水助羧基内质子迁移和质子以新羰基氧为桥从手性碳向苯环迁移的共同历程后,再分别直接迁移到手性碳的另一侧和以新羰基氧为桥迁移到手性碳的另一侧;b是水助质子以羰基氧为桥从手性碳的一侧迁移到另一侧.势能面计算表明,a2是主反应通道,在2个水分子助质子迁移反应时,决速步吉布斯自由能垒被降到最低值124.3kJ·mol-1,与裸反应、限域在MOR分子筛和限域在水环境的此通道决速步能垒287.1,263.4kJ·mol-1和152.2kJ·mol-1相比较,均有明显降低.结果表明: 水与MOR分子筛复合环境对布洛芬手性转变具有较好的共催化作用,可作为理想的实现布洛芬手性转变的纳米反应器.
MOR分子筛; 布洛芬; 手性转变; 密度泛函; 过渡态
布洛芬(Ibuprofen,Ibu)具有手性,有左布洛芬(S-Ibu)和右布洛芬(R-Ibu)两种异构体.医学上广泛使用Ibu治疗强直脊椎炎、风湿关节炎和神经炎等.人们对Ibu已经进行了广泛的研究,文献[1]进行了外消旋Ibu的动力学拆分模拟和实验的研究.文献[2-4]的研究表明,R-Ibu的活性是S-Ibu的160倍,其疗效和安全性也远远优于S-Ibu,在生命体内可以缓慢实现S-Ibu的旋光异构.
文献[5-6]的研究表明,Ibu分子的手性转变裸反应有8条路径,裸反应决速步质子迁移反应的能垒是287.1kJ·mol-1,2个水分子助决速步质子迁移反应的能垒为152.2kJ·mol-1.文献[7]的研究表明,扶椅型SWCNT对Ibu分子手性转变的限域影响是,随着SWCNT直径的减小,手性转变反应路径由2条变为1条,决速步能垒逐渐变小.
目前临床上使用的Ibu多数为消旋体,单一手性的R-Ibu的价格及其昂贵[8],寻找一个既经济又环保的方法使S-Ibu转化为R-Ibu十分重要.分子筛的价格低廉和绿色环保,使利用分子筛实现对Ibu分子手性转变反应的催化具有实际意义.只是利用MOR分子筛实现布洛芬的手性转变反应还不具有可行性.文献[9-10]研究了α-丙氨酸限域在SWBNNT(9,9)和SWBNNT(10,6)分别与水构成的复合环境下的手性转变机制,研究结果表明,纳米管的限域和水分子的共催化作用使反应能垒明显降低.基于文献[9-10]的研究经验,本工作研究了全硅MOR分子筛与水的复合环境对Ibu分子手性转变反应的共催化机制.
1 模型的选取与计算研究方法
MOR分子筛含有12元环和8元环两种一维主直孔道,12元环窗口直径为0.65nm×0.70nm,八元环窗口直径为0.26nm×0.57nm.Ibu与3个水分子以氢键形成的水合分子的最大横向线度为0.60nm×0.53nm,只能进入12元环孔道,因此,将MOR分子筛的12元环孔道作为限域环境.Ibu与3个水分子以氢键形成的水合分子的最大纵向线度为1.369nm,为充分地考虑孔道限域效应,采用周期性模型把含有12元环和部分8元环直孔道分子筛骨架包括进来,用含有240T的簇模型作为限域催化环境(此模型的长度是3.2nm左右),用氢原子饱和模型截断处的硅原子,并把硅氢键长固定为1.46nm,如图1(a)所示.
采用QM/MM组合的ONIOM(Our Own N-layered Integrated molecule Orbit and Molecule Mechanics)方法[11],研究标题反应机制.将分子筛与其内部反应物的水合物等形成的包结物分为两层处理: 内层反应底物为QM区,考虑到分子筛与内部反应底物的长程作用,用CAM(Coulomb-attenuated hybrid exchange-correlation functional)结合DFT的长程校正泛函CAM-B3LYP[12-13]方法,基组采用6-31G(d,p);外层分子筛为MM区,采用分子力学的UFF(Universal Force Field)力场[14]处理,为不使分子筛骨架形变,把外层固定,全参数优化稳定点和过渡态[15-16].对QM区采用6-311++G(2df,pd)基组计算各包结物的高水平单点能,利用Gtotal=ESP+Gtc(ESP和Gtc分别为高水平的单点能和吉布斯自由能热校正)计算高水平的总吉布斯自由能,绘制反应过程的吉布斯自由能势能面.通过对过渡态进行频率分析和内禀反应坐标(IRC)计算[17],确认过渡态的可靠性.Ibu分子与1个水分子以氢键形成的复合分子限域在MOR分子筛内的包结物记为Ibu·1H2O@MOR,其它体系的表示类似.文中计算均采用Gaussian 09软件包[18]完成.
2 结果与讨论
在B3LYP/6-31+G(d,p)水平,优化的单体S型和R型Ibu的几何构型[5],见图1(b)和(c).
通过对图1的分析与计算研究发现,限域在MOR分子筛与水复合环境下,S-Ibu的手性转变反应分为3个通道a1,a2和b.a1和a2的前三步基元反应相同,是水助羧基内质子迁移,形成新的羰基,然后水助质子以新的羰基氧为桥从手性碳迁移到苯环.a1的后续过程是,水助质子从苯环直接迁移到手性碳的另一侧,实现手性转变;a2的后续过程是,水助质子以新羰基氧为桥从苯环迁移到手性碳的另一侧,实现手性转变.b通道是水助手性碳上的质子直接以羰基氧为桥,从手性碳的一侧迁移到另一侧,实现手性转变.水环境下,可以有很多水分子以氢键的形式与反应过程中的驻点形成水合分子,计算表明,质子迁移反应能垒只与参与反应的水分子个数有关.因此,我们只讨论参与反应的水分子个数不同的情形.研究发现,由于分子筛12元环孔道的限制,只能是1,2个和3个水分子作水助质子迁移反应的媒介.下面对3个通道上的反应机理分别进行讨论.
2.1 限域在MOR分子筛与水复合环境下S-Ibu在a1和a2通道的手性转变反应机理
1水助a1和a2的反应历程见图2A(第742页),共同的反应历程见图2A的X,篇幅所限只对前2个基元反应机理做详细讨论.首先是第1基元反应,羧基内质子迁移.S-Ibu与羧基右侧的1个水分子以氢键结合的水合分子的包结物a1(a2)S-Ibu·1H2O@MOR,经过1水助羧基内氢迁移的6元环过渡态a1(a2)STS1·1H2O@MOR,异构成中间体产物包结物a1(a2)SINT1·1H2O(m)@MOR(此处m表示水分子在羧基右侧,后面雷同的表示不再予以解释),738O成为新羰基.此过程反应物S-Ibu·1H2O的741H与742O的距离是0.17078nm,743H与739O的距离是0.19087nm,水分子与S-Ibu在羧基的方向以强氢键结合.过渡态STS1·1H2O的氢键键角738O-741H-742O和742O-743H-739O都是153.5°,距离平角180.0°相差很大,氢键较弱,这说明六元环结构过渡态STS1·1H2O不稳定,会产生较大的能垒.接着是第2基元反应,质子从手性碳向新羰基的迁移过程.SINT1与其740H和738O前面的1个水分子以氢键结合的水合分子的包结物a1(a2)SINT1·1H2O(n)@MOR(此处n表示水分子在740H和738O的前面,后面雷同的表示不再予以解释),经1个水分子助手性碳上的H向新羰基O迁移的6元环过渡态a1(a2)TS2·1H2O@MOR,异构成中间体产物包结物a1(a2)INT2·1H2O(m)@MOR,此时INT2的羧基质子化.TS2·1H2O的氢键键角720C-740H-742O和742O-743H-738O分别是149.8°和154.3°,距离平角180.0°相差更大,氢键更弱,这说明六元环结构过渡态TS2·1H2O更不稳定,会产生更大的能垒;反应物SINT1·1H2O(n)的740H与手性碳720C断键需要的能量也要高于S-Ibu·1H2O的741H-738O键断需要的能量.因此,第2基元反应的能垒高于第1基元反应,后面的势能面计算结果说明此分析是正确的.第3基元反应是INT2与苯环和743H前面的1个水分子以氢键结合的水合分子包结物a1(a2)INT2·1H2O(n)@MOR,经过以水分子为媒介,质子化羧基上的743H向苯环迁移的过渡态a1(a2)TS3·1H2O@MOR,异构成中间体包结物a1(a2)INT3·1H2O(m)@MOR.TS3·1H2O的氢键键角分别是155.9°和166.8°,比TS2·1H2O的大些,因此,过渡态a1(a2)TS3·1H2O@MOR会比a1(a2)TS2·1H2O@MOR稳定.又由于反应物a1(a2)INT2·1H2O(n)@MOR到a1(a2)TS3·1H2O@MOR是断H-O键,比断H-C键容易.因此,a1(a2)TS3·1H2O@MOR产生的能垒会比a1(a2)TS2·1H2O@MOR低很多.
从INT3开始分成2个路径,a1路径的反应历程见图2A的Y.INT3与苯环和手性碳后面的1个水分子以氢键结合的水合分子的包结物a1INT3·1H2O(n1)@MOR,经1水助质子从苯环到手性碳的过渡态a1TS4·1H2O@MOR,异构成产物a1P1R-Ibu·1H2O(m)@MOR,完成手性转变.经过以1个水分子为媒介质子在羧基内回迁的过渡态a1RTS5·1H2O@MOR,a1P1R-Ibu·1H2O(n1)@MOR异构成为产物a1P2R-Ibu·1H2O@MOR.a2路径的反应历程见图3A(第745页)的Z.先是INT3与苯环和新羰基之间的1个水分子以氢键结合的水合分子的包结物a2INT3·1H2O(n2)@MOR,经1个水分子助质子从苯环向新羰基迁移的过渡态a2TS4·1H2O@MOR,异构成产物中间体a2INT4·1H2O(m)@MOR.接着INT4与其手性碳和质子化羧基之间的1个水分子以氢键结合的水合分子a2INT4·1H2O(n)@MOR,经质子从质子化羧基向手性碳迁移的过渡态a2TS5·1H2O@MOR,异构成产物a2P1R-Ibu·1H2O@MOR,完成手性转变.最后,a2P1R-Ibu·1H2O(m)@MOR,经过与a1RTS5·1H2O@MOR雷同的过渡态a2RTS5·1H2O@MOR,异构成产物a2P2R-Ibu·1H2O@MOR.此基元反应的各驻点包结物构象与a1第5基元反应的基本相同,从略.
2个和3个水分子助S-Ibu的手性转变历程同于1个水分子的情形,计算表明,第2步基元反应是决速步,为节省篇幅,只讨论前2步基元反应历程.
2水助a1和a2共同的前2步反应历程见图2B(第743页). S -Ibu与羧基右侧的2个水分子以氢键结合的水合分子的包结物a1(a2)S -Ibu·2H2O@MOR,经过2水助羧基内氢迁移的8元环过渡态a1(a2)STS1·2H2O@MOR,异构成中间体产物包结物a1(a2)SINT1·2H2O(m)@MOR. SINT1与其手性碳上的740H和新羰基738O前面的2个水分子以氢键结合的水合分子的包结物a1(a2)SINT1·2H2O(n)@MOR,经2个水分子助手性碳上的H向新羰基O迁移的8元环过渡态a1(a2)TS2·2H2O@MOR,异构成中间体产物包结物a1(a2)INT2·2H2O(m)@MOR. 3水助a1和a2共同的前2步反应历程见图2C(第744页). S -Ibu与羧基右侧的3个水分子以氢键结合的水合分子的包结物a1(a2)S -Ibu·3H2O@MOR,经过3水助羧基内氢迁移的10元环过渡态a1(a2)STS1·3H2O@MOR,异构成中间体产物包结物a1(a2)SINT1·3H2O(m)@MOR.SINT1与其手性碳上的740H和新羰基738O前面的3个水分子以氢键结合的水合分子的包结物a1(a2)SINT1·3H2O(n)@MOR,经3个水分子助手性碳上的H向新羰基O迁移的过渡态a1(a2)TS2·3H2O@MOR,异构成中间体产物包结物a1(a2)INT2·3H2O(m)@MOR.分子结构计算表明: a1(a2)TS2·2H2O@MOR的氢键键角720C-740H-742O,742O-743H-745O和745O-747H-738O分别是167.04°,166.62°和168.94°,a1(a2)TS2·3H2O@MOR的氢键键角720C-740H-742O、742O-743H-748O、748O-749H-745O和745O-747H-738O分别是167.80°,172.78°,171.80°和174.33°,均比a1(a2)TS2·1H2O@MOR中的氢键键角153.5°增大许多,更接近180.00°.这导致8元环结构a1(a2)TS2·2H2O@MOR和10元环结构a1(a2)TS2·3H2O@MOR均比6元环结构a1(a2)TS2·1H2O@MOR稳定许多,因此,过渡态a1(a2)TS2·2H2O@MOR和a1(a2)TS2·3H2O@MOR产生的能垒比a1(a2)TS2·1H2O@MOR低.a1(a2)TS2·3H2O@MOR的10元环结构与8元环结构a1(a2)TS2·2H2O@MOR相比较,对应的氢键键角略增大,似乎应该结构更稳定,产生的能垒会略低些.但过渡态a1(a2)TS2·3H2O@MOR的10元环结构图和二面角数据显示,过渡态a1(a2)TS2·3H2O@MOR的10元环结构明显偏离平面,这说明其结构不稳定,会导致其产生的能垒增高.因此,10元环结构过渡态a1(a2)TS2·3H2O@MOR与8元环结构过渡态a1(a2)TS2·2H2O@MOR产生的能垒不会低,甚至可能高些,后面的势能面计算证明了此处分析的正确性.
在ONIOM(CAM-B3LYP/6-311++G(2df,pd): UFF)∥ONIOM(CAM-B3LYP/6-31G(d,p): UFF)双理论水平,全参数优化a1和a2路径上的各个驻点包结物,计算单点能.驻点包结物的几何结构和过渡态在虚频下的振动模式见图2.对诸过渡态进行的频率分析和IRC计算,确认了过渡态的可靠性.各驻点包结物的吉布斯自由能热校正和过渡态虚频(Ima)见表1.驻点包结物的单点能,热校正总吉布斯自由能和相对总吉布斯自由能见表1(第744~745页).
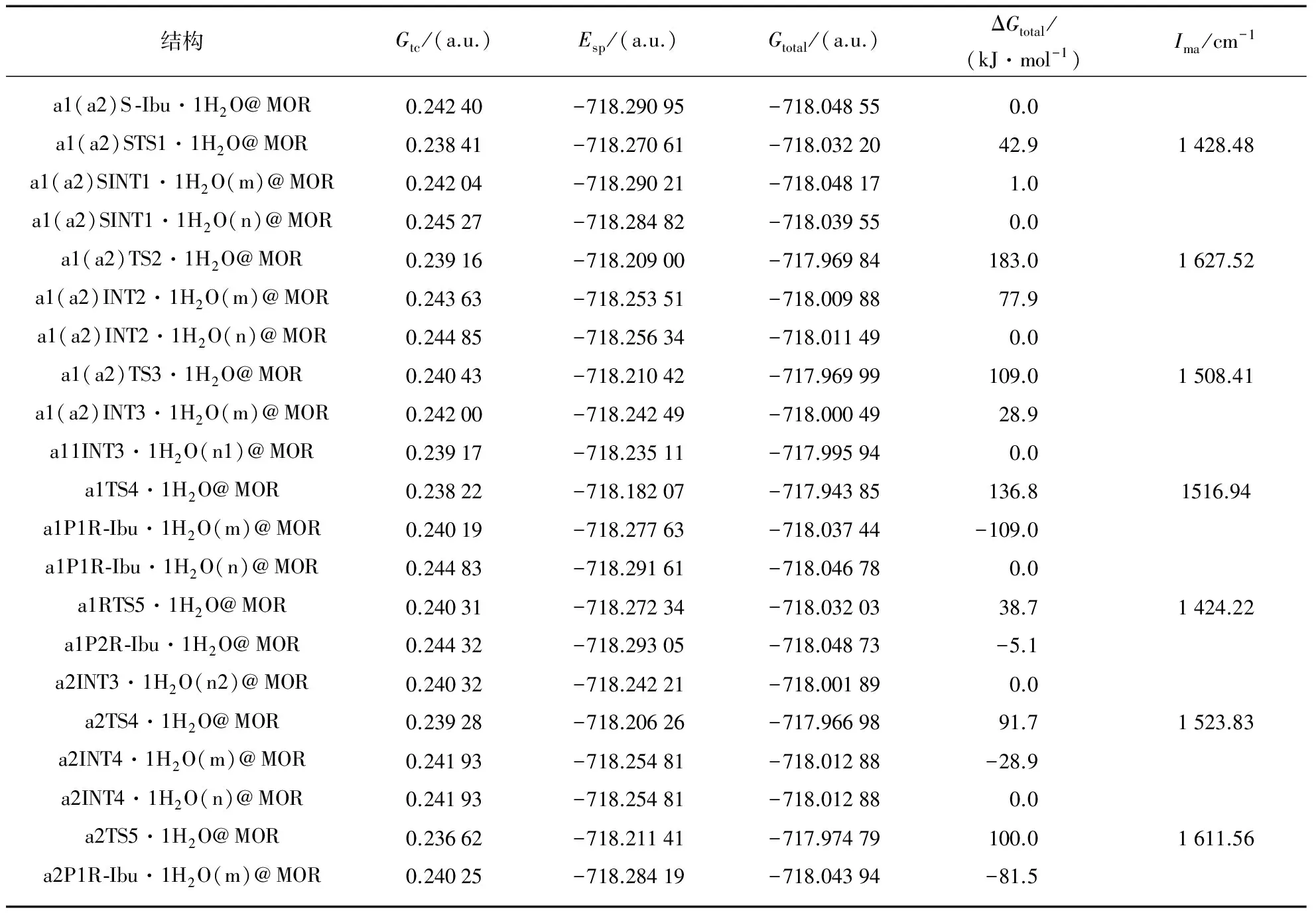
结构Gtc/(a.u.)Esp/(a.u.)Gtotal/(a.u.)ΔGtotal/(kJ·mol-1)Ima/cm-1a1(a2)S⁃Ibu·1H2O@MOR0.24240-718.29095-718.048550.0a1(a2)STS1·1H2O@MOR0.23841-718.27061-718.0322042.91428.48a1(a2)SINT1·1H2O(m)@MOR0.24204-718.29021-718.048171.0a1(a2)SINT1·1H2O(n)@MOR0.24527-718.28482-718.039550.0a1(a2)TS2·1H2O@MOR0.23916-718.20900-717.96984183.01627.52a1(a2)INT2·1H2O(m)@MOR0.24363-718.25351-718.0098877.9a1(a2)INT2·1H2O(n)@MOR0.24485-718.25634-718.011490.0a1(a2)TS3·1H2O@MOR0.24043-718.21042-717.96999109.01508.41a1(a2)INT3·1H2O(m)@MOR0.24200-718.24249-718.0004928.9a11INT3·1H2O(n1)@MOR0.23917-718.23511-717.995940.0a1TS4·1H2O@MOR0.23822-718.18207-717.94385136.81516.94a1P1R⁃Ibu·1H2O(m)@MOR0.24019-718.27763-718.03744-109.0a1P1R⁃Ibu·1H2O(n)@MOR0.24483-718.29161-718.046780.0a1RTS5·1H2O@MOR0.24031-718.27234-718.0320338.71424.22a1P2R⁃Ibu·1H2O@MOR0.24432-718.29305-718.04873-5.1a2INT3·1H2O(n2)@MOR0.24032-718.24221-718.001890.0a2TS4·1H2O@MOR0.23928-718.20626-717.9669891.71523.83a2INT4·1H2O(m)@MOR0.24193-718.25481-718.01288-28.9a2INT4·1H2O(n)@MOR0.24193-718.25481-718.012880.0a2TS5·1H2O@MOR0.23662-718.21141-717.97479100.01611.56a2P1R⁃Ibu·1H2O(m)@MOR0.24025-718.28419-718.04394-81.5
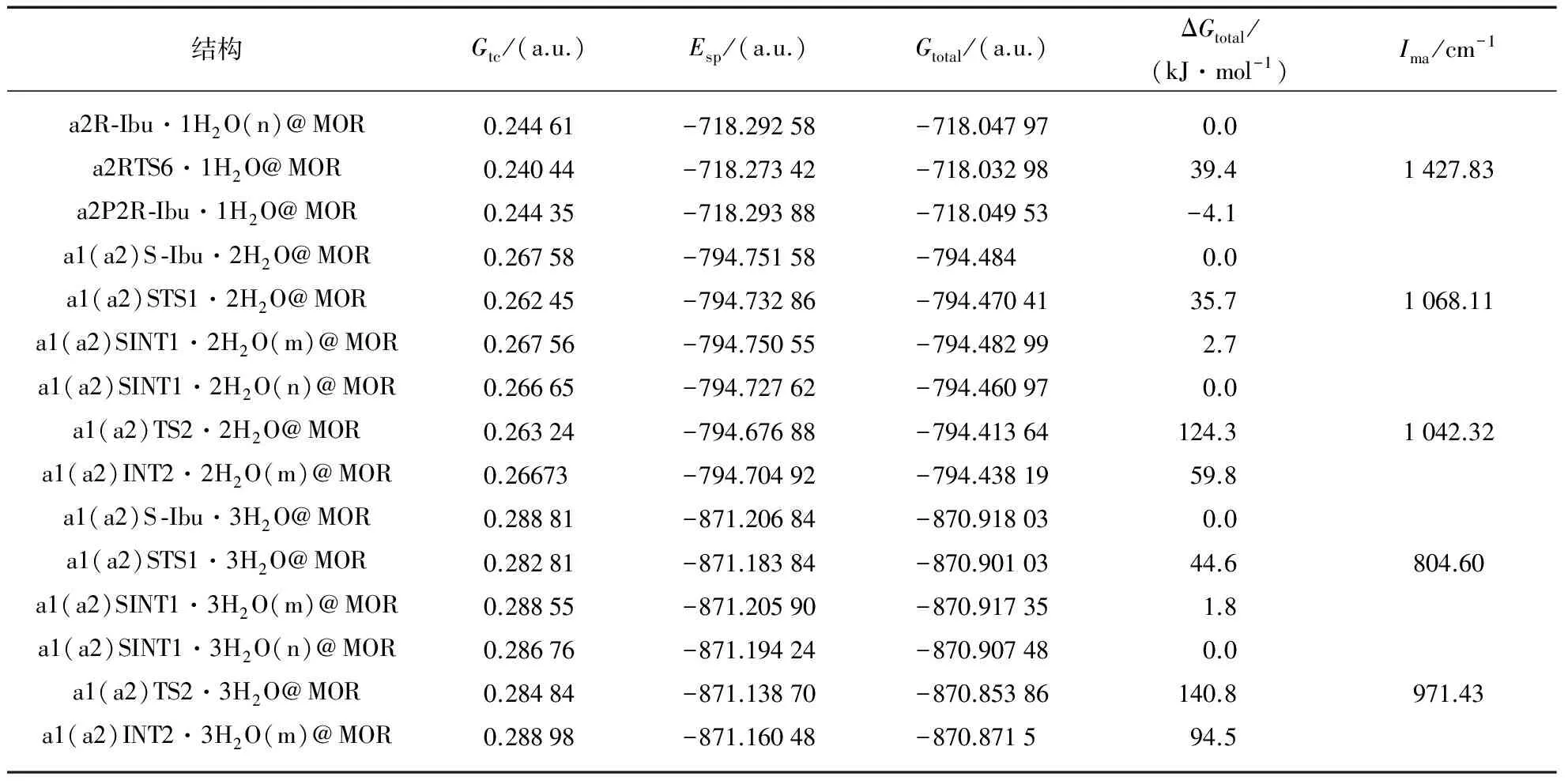
(续表)
根据表1中的数据,绘制了标题反应在a1和a2反应通道上的吉布斯自由能势能面,见图3.
从图3看出,标题反应在a1和a2通道分别经过5个和6个过渡态,通过5个和6个基元反应实现,前3个基元反应是共同的过程.第2步基元反应为共同的决速步骤,对于第4基元反应,a2通道的能垒明显低,为优势反应通道.a1和a2通道的决速步能垒在2个水分子作氢迁移媒介时被降到最低值124.3kJ·mol-1,是由手性碳上的H直接向新羰基氧738O迁移的过渡态产生的.比裸反应和限域在水环境的此通道决速步能垒287.1和152.2kJ·mol-1[5-7]相比较大幅度降低或明显降低.说明MOR分子筛与水的复合环境对S -Ibu在a1和a2通道的旋光异构具有显著的限域共催化作用.
2.2 限域在MOR分子筛与水复合环境下的S -Ibu在b通道手性转变决速步的反应机理
S -Ibu在b通道手性转变反应决速步见图4.S -Ibu与手性碳、手性碳上的质子和羰基前面的1个、2个和3个水分子以氢键结合的水合分子的包结物bS -Ibu·1H2O@MOR、bS -Ibu·2H2O@MOR和bS -Ibu·3H2O@MOR,分别经过图4所示的六元环、八元环和十元环结构过渡态bTS1·1H2O@MOR、bTS1·2H2O@MOR和bTS1·3H2O@MOR,异构成产物中间体bINT1·1H2O@MOR、bINT1·2H2O@MOR和bINT1·3H2O@MOR.以后的过程先是,bINT1考虑溶剂效应的羧基异构.bINT1经过质子化羧基上的H在纸面里外反转的过渡态,质子化羧基上的1个质子H摆到纸面里(此过程能垒很小).然后,摆到纸面里的质子再以水分子为媒介在纸面里迁移到手性碳,完成手性转变过程,此过程雷同于第1基元反应.篇幅所限,不对这两个基元反应进行讨论.
过渡态的结构数据显示: bTS·1H2O@MOR的氢键键角在149.9°~154.9°之间,bTS·2H2O@MOR和bTS·3H2O@MOR氢键键角在169.9°~170.8°和169.3°~173.6°之间,明显大于前者,更接近180°.所以bTS·2H2O@MOR和bTS·3H2O@MOR的结构比bTS·1H2O@MOR稳定,产生的能垒要低些.bTS·2H2O@MOR和bTS·3H2O@MOR的结构数据显示,过渡态bTS·3H2O@MOR十元环结构与过渡态bTS·2H2O@MOR的八元环结构相比较,明显偏离平面.说明过渡态bTS·3H2O@MOR相对不稳定,产生的能垒会相对高,这抵消了bTS·3H2O@MOR比bTS·2H2O@MOR的氢键稍强的因素,使得他们二者产生的能垒不会相差很大,后面的势能面计算说明了此分析的正确性.
在与前面相同的理论计算水平上,全参数优化b通道上不同个数的水分子助决速步骤的驻点包结物,计算单点能.得到b通道各驻点包结物的几何构型以及过渡态在虚频下的振动模式,见图4.对诸过渡态的频率分析和IRC计算,确认了过渡态的可靠性.各驻点吉布斯自由能热校正和过渡态虚频(Ima)见表2.驻点的高水平单点能,热校正的总自由能和相对总自由能亦见表2.

结构Gtc/(a.u.)Esp/(a.u.)Gtotal/(a.u.)ΔGtotal/(kJ·mol-1)Ima/cm-1bS⁃Ibu·1H2O@MOR0.24049-718.28229-718.04180.0bTS·1H2O@MOR0.23912-718.21208-717.97296180.71724.19bINT1·1H2O@MOR0.24211-718.24800-718.0058994.3bS⁃Ibu·2H2O@MOR0.26489-794.73339-794.46850.0bTS·2H2O@MOR0.25977-794.67297-794.4132145.21388.65bINT1·2H2O@MOR0.26388-794.70321-794.4393376.6bS⁃Ibu·3H2O@MOR0.28568-871.18914-870.903460.0bTS·3H2O@MOR0.28389-871.13217-870.84828144.9967.77bINT1·3H2O@MOR0.28958-871.16481-870.8752374.1
根据表2的数据,绘制了Ibu限域在MOR分子筛,在b通道水助手性转变决速步反应过程的吉布斯自由能势能面示意图,见图5.
从图5可以看出,标题反应在b通道的决速步骤能垒在以2个和3个水分子为质子迁移媒介时被降到最低值145.2和144.9kJ·mol-1,是由质子H从手性碳向羰基氧739O迁移的过渡态产生的.比裸反应和只限域水环境的决速步能垒295.8和170.4kJ·mol-1[5-6]有明显降低和大幅度地降低.说明MOR分子筛对S -Ibu在b通道的旋光异构具有较好的限域催化作用.与前面2.1的研究结果共同说明,MOR分子筛与水的复合环境可以作Ibu旋光异构反应的理想纳米反应器.综合图3和5可知,a2是主反应通道,在2个水分子助质子迁移反应时,决速步吉布斯自由能垒被降到最低值124.3kJ·mol-1.
分子结构研究表明: 2个和3个水分子助氢迁移反应的过渡态分子氢键键角比1个水分子助氢迁移反应时明显增大,3个水分子助氢迁移反应的10元环过渡态结构明显偏离平面.反应通道研究发现: 标题反应有a1、a2和b三个通道.a1和a2是经过水助羧基内质子迁移和质子以新羰基氧为桥从手性碳向苯环迁移的共同历程后,再分别直接迁移到手性碳的另一侧和以新羰基氧为桥迁移到手性碳的另一侧,要分别经过5个和6个基元反应;b是水助质子以羰基氧为桥从手性碳的一侧迁移到另一侧,要经历3个基元反应.反应势能面计算表明,a2是主反应通道,在2个水分子助质子迁移反应时,决速步吉布斯自由能垒被降到最低值124.3kJ·mol-1,与裸反应、只是限域在MOR分子筛和限域在水环境的此通道决速步能垒287.1,263.4kJ·mol-1和152.2kJ·mol-1相比较,分别大幅降低和明显降低.结果表明: 水与MOR分子筛复合环境对布洛芬分子的手性转变具有较好的限域共催化作用,可作为实现布洛芬手性转变的理想的纳米反应器.
用铝取代某位置的硅形成B酸位,此时酸催化的作用可能更值得期待,此工作正在进行中.
[1] BHATIA S, LONG W S, KAMARUDDIN A H. Enzymatic membrane reactor for the kinetic resolution of racemic ibuprofen ester: modeling and experimental studies[J].ChemicalEngineeringScience, 2004,59(22-23): 5061-5068.
[2] 肖方清.右旋布洛芬的制备[J].中国医药工业杂志,2000,31(11): 486-488.
[3] 林文辉.手性药物布洛芬的体内药物动力学研究[D].沈阳: 沈阳药科大学,2004: 8-10.
[4] CHENG H, ROGERS J D, DEMETRIADES J L,etal. Pharmacokinetics and bioinversion of ibuprofen enantiomers in humans[J].PharmaceuticalResearch, 1994,11(6): 824-830.
[5] 邹晓威,梅泽民,王佐成,等.孤立条件下布洛芬分子手性转变过程的理论研究[J].原子与分子物理学报,2015,32(2): 173-180.
[6] 梅泽民,王佐成,闫红彦,等.水环境下布洛芬分子的手性转变机理[J].吉林大学学报(理学版).2015,53(2): 331-339.
[7] 王佐成,梅泽民,吕 洋,等.扶手椅型单壁碳纳米管的尺寸对布洛芬分子手性转变的限域影响[J].复旦学报(自然科学版).2015,54(2): 234-244.
[8] 赵亚华.分子生物学教程[M].北京: 科学出版社,2011: 5-6.
[9] 梅泽民,佟 华,王佐成,等.α-丙氨酸限域在扶椅型SWBNNT(9,9)与水复合环境下的手性转变机制[J].中山大学学报(自然科学版).2016,54(3): 85-92.
[10] 佟 华,梅泽民,王佐成,等.α-Ala限域在螺手性SWBNNT(10,6)与水复合环境下手性转变机理[J].复旦学报(自然科学版).2016,55(4): 529-540.
[11] SVENSSON M, HUMBEL S, FROESE R D J,etal. ONIOM: A multilayered integrated MO+MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2+ H2oxidtivae addition[J].PhysicalChemistry, 1996,100(50): 19357-19363.
[12] KOBAYASHI R, AMOS R D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin-bacteriochlorin complex[J].ChemPhysLetts, 2006,420: 106-109.
[13] YIN S W, DAHLBOM M G, CANFIELD P J,etal.Assignment of the Qy absorption spectrum of photosystem-I from thermosynechococcus elongatus based on CAM-B3LYP calculations at the PW91-optimized protein structure[J].PhysChemB, 2007,111(33) : 9923-9930.
[14] RAPPE A K, CASEWIT C J, COLWELL K S,etal. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations[J].JournaloftheAmericanChemicalSociety, 1992,114(25): 10024-10053.
[15] GARRETT B C, TRUHLAR D G. Generalized transition state theory. Classical mechanical theory and applications to collinear reactions of hydrogen molecules[J].JournalofPhysicalChemistry, 1979,83(8): 1052-1079.
[16] GARRETT B C, TRUHLAR D G. Criterion of minimum state density in the transition state theory of bimolecular reactions[J].TheJournalofChemicalPhysics, 1979,70(4): 1593-1598.
[17] ISHIDA K, MOROKUMA K, KOMORNICKI A. The intrinsic reaction coordinate. An ab initio calculation for HNC→HCN and H-+ CH4→CH4+ H-*[J].TheJournalofChemicalPhysics, 1977,66(5): 2153-2156.
[18] FRISCH M J, TRUCKS G W, SCHLEGEL H B,etal. Gaussian 09. Revision D.01[CP]. Pittsburgh USA: Gaussian, Inc., Wallingford CT, 2013.
Theoretical Research on the Co-catalysis of Water and MOR Zeolite Combined Environment on the Chiral Transition of Ibuprofen Molecules
SUN Yongqing1, WANG Zuocheng2, Gao Feng2, TANG Dehuai3
(1.DepartmentofBasic,BaichengTechnicianInstitute,Baicheng137000,China;2.CollegeofPhysicsandElectronicInformation,BaichengNormalUniversity,Baicheng137000,China;3.ComputerScienceCollege,BaichengNormalUniversity,Baicheng137000,China)
The chiral transition of ibuprofen molecules confined in water and MOR zeolite combined environment was studied by introducing the ONIOM methods using combination of quantum mechanics and molecular mechanics. The molecular structure researches show that hydrogen bond angles of the transition state molecules increases constantly in the hydrogen transfer reactions with the help of one water molecule, two and three molecules, respectively, and the 10-ring transition state structures in the hydrogen transfer reactions with the help of three water molecules significantly deviates from the plane. The study of reaction channels shows there are three channels a1, a2 and b in the title reaction. The protons transfer with the help of water molecules in carboxyl and from the chiral C to the benzene ring using the carboxyl O as the bridge, and then transfer to the other side of the chiral carbon directly or with new carbonyl O as a bridge in the channels a1and a2, respectively. In channel b, the protons transfer with the help of water molecules transfer from one side to the other of the chiral C with carbonyl O as a bridge. Calculations of potential energy surface show a2 channel is the dominant reaction path, where the rate determined step of Gibbs free energy barrier of two water-assisted proton transfers reaction is reduced to the minimum value 124.3kJ·mol-1, which is significantly lower than the Gibbs free energy barrier 287.1kJ·mol-1, 263.4kJ·mol-1and 152.2kJ·mol-1respectively corresponding to the bare reaction, confined in MOR zeolite and confined in water environment. The results show that water and MOR zeolite combined environment has a good co-catalysis on the chiral transition of ibuprofen molecules and can be used as an ideal nano reactor for the realization of ibuprofen molecules chiral transition.
MOR zeolite; Ibuprofen; chiral transition; density functional; transition state
0427-7104(2016)06-0739-11
2016-06-12
吉林省科技发展计划项目(20160101308JC)
孙永清(1968—),男,副教授. E-mail: syfytw@163.com;通讯联系人,王佐成(1963—),男,副教授,E-mail: wangzc188@163.com.
O 641(O641.12;O641.12)
A
猜你喜欢
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
山西大同大学学报(自然科学版)(2022年3期)2022-07-06
黑龙江大学自然科学学报(2022年1期)2022-03-29
煤气与热力(2021年9期)2021-11-06
波谱学杂志(2021年3期)2021-09-07
湖南大学学报·自然科学版(2020年2期)2020-04-17
江苏农业科学(2017年20期)2017-11-30
中学化学(2015年12期)2016-01-19
中学化学(2015年12期)2016-01-19
