
毛细管电泳与化学发光检测联用方法的研究进展
2017-01-09 12:06:36黄香宜任吉存上海交通大学化学化工学院金属基复合材料国家重点实验室上海200240
色谱 2017年1期
易 方, 黄香宜, 任吉存(上海交通大学化学化工学院, 金属基复合材料国家重点实验室, 上海 200240)
邹汉法研究员纪念专辑(下)·专论与综述
毛细管电泳与化学发光检测联用方法的研究进展
易 方, 黄香宜*, 任吉存*
(上海交通大学化学化工学院, 金属基复合材料国家重点实验室, 上海 200240)
毛细管电泳由于其超高的分离效率广泛应用于生物医药、环境监测、食品科学以及公共安全等领域。然而,由于毛细管电泳具有进样量较少、检测光程较短等缺点,需要与高灵敏度检测器联用实现低浓度样品的分析。化学发光检测由于其背景信号低而具有超高的灵敏度。毛细管电泳-化学发光检测联用方法将毛细管电泳的高效分离特性与化学发光检测的高灵敏性相结合,成为一种非常重要的分析方法,广泛用于化学分析、药物筛选以及环境监测等领域。该文对近年来毛细管电泳-化学发光检测联用方法的基本原理进行概述,并对其发展趋势和应用前景进行了展望。
毛细管电泳;化学发光检测;综述
毛细管电泳(capillary electrophoresis, CE)是一类以毛细管为分离通道、以高压直流电场为驱动力的新型液相分离分析技术。CE迅速发展于20世纪80年代后期,是分析科学中继高效液相色谱之后的又一重大进展,是最有效的分析方法之一。它使分析科学得以从微升水平进入纳升水平,并使单细胞乃至单分子分析成为可能。芯片毛细管电泳(microchip capillary electrophoresis, MCE)是在常规毛细管电泳原理和技术的基础上,利用微加工技术在平方厘米级大小的芯片上加工出各种微细结构,通过不同的通道、反应器、检测单元等设计和布局,实现样品进样、反应和分离检测的微型实验装置。
CE和MCE相对于常规分析方法具有分析时间短、样品及试剂用量少和分离效率高等特点,已广泛用于化学和生物分析、药物分析、环境以及食物分析等领域。然而,极少的样品量对检测灵敏度提出了更高的要求。对CE和MCE而言,选择合适的检测器是非常重要的。紫外可见吸收检测法具有良好的通用性,通常作为商品化毛细管电泳仪的常规检测器。但是紫外可见吸收检测法灵敏度较低,加上毛细管内径细小、检测光程短等不足,导致该检测方法的灵敏度不能满足某些低含量生物样品检测的要求[1]。目前,某些更灵敏的检测方法得以发展,比如激光诱导荧光检测[2-4]、电化学检测[5,6]、质谱检测[7]等。其中激光诱导荧光检测和质谱检测具有高灵敏度,但设备成本较高,使其应用受到一定的限制;电化学检测具有高灵敏度且成本较低,但具有一些如电极表面污染等应用限制[8]。因此,发展高灵敏、高选择性的微体积检测方法非常重要。
化学发光(chemiluminescence,CL)检测方法是指在没有外加光源的情况下,由化学反应过程提供能量,从而产生光辐射来进行定量分析的方法。CL检测方法因其分析速度快、线性范围宽、仪器设备简单及易实现自动化等特点,成为痕量分析领域的研究热点之一。与其他的光学检测方法相比,CL检测由于没有来自光源杂散光的干扰,可获得极低的检测噪声,因此被认为是最灵敏的检测方法之一[9]。CE-CL联用方法实现了CE高效分离与CL高灵敏度检测的结合,并具有以下优点[10]: (1)背景信号低,检出限可达到单分子水平,且具有与激光诱导荧光检测方法相当的高灵敏度;(2)具有较宽的线性范围,有利于对分析物进行定量分析;(3)不需要光源,仪器的配制更简单。
CE-CL联用方法已经成功应用于无机和有机化合物的定量分析,主要包括金属离子、药物、氨基酸、核酸以及蛋白质的分析研究[11]。本文对近年来CE-CL联用方法的研究和发展进行了总结与展望。
1 CE-CL联用的发光体系
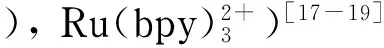
1.1 鲁米诺体系
到目前为止,luminol-CL体系在CE-CL检测系统中应用最为广泛,已用于金属离子、有机小分子、糖类和蛋白质等物质的检测分析。Albrecht[13]早在1928年提出luminol及其衍生物在碱性溶液(pH=10~11)中加入H2O2时可以观测到微弱的蓝色发光(发射波长425~435 nm)现象。加入适当的催化剂,如Co(Ⅱ)、Fe(Ⅱ)、Mn(Ⅱ)、Cu(Ⅱ)、辣根过氧化物酶(horseradish peroxidase,HRP)等,可以极大地提高CL强度。H2O2、K3[Fe(CN)6]、NaClO、KIO4和KMnO4等氧化剂可以诱导luminol反应并产生CL信号[20]。在该体系中,luminol与氧化剂在碱性条件下反应,而大部分金属离子在碱性环境中会生成氢氧化物沉淀,失去对luminol的催化作用。为了避免沉淀,可以将碱性电泳缓冲液换成酸性电泳缓冲液,之后再增加pH值。Zhang等[21]将这种方法用于分析异烟肼和对氨基水杨酸。这两种分析物在luminol-H2O2反应体系中可以提高Cu2+的催化能力,增强的CL信号分别与这两种分析物的浓度成比例。
K3[Fe(CN)6]在luminol反应中也是合适的氧化剂和催化剂。在luminol-K3[Fe(CN)6]反应体系中,Han等[22]利用米托蒽醌在CL反应中的增敏作用提高了分析方法的灵敏度,将该方法成功用于人血浆和尿液样品中米托蒽醌的检测。进而,该课题组[23]证明了在没有任何催化剂的条件下,烟酰胺可以有效抑制luminol-K3[Fe(CN)6]体系的发光强度,并通过CE-CL联用方法快速、可靠、灵敏地对烟酰胺进行微量检测,成功应用于食品、血浆样品中的检测。Zhu等[24]发现KIO4在K3[Fe(CN)6]的催化下可以快速与luminol发生反应,luminol-KIO4-K3[Fe(CN)6]反应体系在0.65 s内完成CL反应。Mu等[25]利用甲状腺素(thyroxine,T4)在碱性条件下对luminol-KMnO4反应体系CL信号的增强作用,检测了正常人与甲亢患者以及甲状腺功能衰退患者血清中T4的含量。结果表明,甲亢患者血清中T4的含量高于正常人的含量,而甲状腺功能衰退者血清中T4的含量低于正常人的含量。因此,该方法可以快速地对甲亢患者或甲状腺功能衰退患者进行初步诊断。
1.2 PO体系
PO反应中,PO在荧光基团的存在下被H2O2氧化,生成一种双氧基中间体储能物,随后发生能量转移至荧光基团,该荧光基团发出高量子产率的荧光[8]。荧光标记方法很容易应用于PO-CL反应体系中,因此利用该体系可以检测荧光标记的化合物[26]。PO在高效液相色谱中已经广泛应用,但由于其在水溶液中溶解度较低且不稳定,故在CE的应用中受到一定限制[20]。Zhu等[27]提出了基于双(2,4,6-三氯苯基)草酸酯-H2O2反应体系的CE-CL检测系统。该系统可以在7 min内对罗丹明6G(R6G)及其水解产物R6G-COOH进行有效分离,对R6G的碱性水解过程进行监测并对速率常数和活化能进行估算。Tsuge等[26]在CE-CL检测体系中同时引入luminol和PO,对异硫氰酸荧光素、异鲁米诺异硫氰酸盐以及这两种试剂标记的蛋白质进行分析。
1.3 吖啶酯体系
吖啶类衍生物具有很高的CL量子产量以及超低的背景信号,且衍生化过程简单、快速,常用于免疫分析。通常采用的体系是吖啶酯-H2O2体系,即用吖啶酯或吖啶磺酰胺标记抗原或抗体,用HNO3、H2O2和NaOH作为发光启动试剂。Guo等[28]采用吖啶酯标记的多肽和蛋白质来测定刻蚀的毛细管接口孔径。Wang等[16]通过CE-CL联用方法实现了人尿液中多巴胺和西布特罗的分离检测。该方法采用吖啶酯作为衍生化试剂,分析过程在400 s之内完成,检出限分别为2.0和0.50 μg/L。
1.4 直接氧化体系
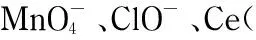
1.5 钌(Ⅱ)联吡啶体系
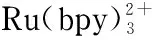
2 CE-CL检测模式
由于不需要光源,CE-CL及MCE-CL检测系统的结构相对简单,检测部分像常规CL测试方法一样仅由一个光电倍增管组成。然而,把CL检测应用于CE或MCE系统中最大的挑战是如何在线引入一种或两种CL试剂。因此,构建适合的CL试剂引入和检测界面对CE-CL检测系统的灵敏度和分离效率具有重要影响。一般按照检测窗口和接地电极相对位置的不同,将接口分为在柱检测模式、离柱检测模式和柱端检测模式等。
在柱检测模式[37,38]由于其较高的灵敏度和分离效率成了CE-CL系统常用的检测模式。在柱式接口大都采用套管式设计,即分离毛细管的末端插入内径稍大的反应毛细管中。1991年,Hara等[39]首次将在柱式接口与CE-CL联用,Dadoo等[40]随后对这一体系进行了系统的研究。Whang等[41,42]所用的接口有所不同,分离毛细管的末端不需要氢氟酸刻蚀,直接插入反应毛细管中,在反应毛细管上烧刻光窗即可。由于该模式的检测窗口一般位于电泳电路之间,因此在高压下不稳定的发光试剂不适用于该装置。为了解决这一问题,Jiang等[43]对蓄液池进行改进,将运行缓冲池和电解池用电介质陶瓷连接在一起,实现了对烟草样品中绿原酸和芦丁含量的检测。
离柱检测模式[44,45]的检测窗口远离电泳高压电源,位于接地电极的后方,主要采用的是柱后套管式。柱后套管式与在柱套管式类似,均将分离毛细管插入内径稍大的反应毛细管中,但是检测窗口和接地电极的相对位置不同。为了解决CL试剂受电压影响的问题,Lee等[44]在毛细管上设计一个断口,用醋酸纤维酯覆盖形成多孔聚合物接点并插入一个装满缓冲液且带有电极的瓶子内,实现了电泳高压分离区与CL检测区的分离。Lin等[46]将离柱式接口置于光电倍增管(photomultiplier tube, PMT)前方,接口由两块不同厚度的有机玻璃板和一些不同大小的分离、反应毛细管组成。该方法成功应用于4种血色素蛋白质的分离及检测,并实现了对人尿液样品中肌红蛋白的测定。
柱端检测模式[47,48]实际是在柱检测模式的一种,最大的特点是缓冲液、发光试剂、接地电极、检测窗口及分离毛细管出口集中于一个储液池内。这种接口的接地电极与检测窗口的相对位置重合。虽然该模式搭建简单且具有较高的灵敏度,但试剂更换较为频繁。另外,由于CL试剂是静止的,导致分析物的峰形拖尾和展宽从而使分离效率降低。Wang等[49]提出了一种旋转式反应池(见图1)。池内有一个石英底的圆形窄通道用于盛CL试剂,相当于一个反应槽。分离毛细管位于反应槽底部附近,这样分离出来的样品可以与CL试剂发生反应。PMT位于反应通道的下方以检测CL信号。该实验采用HRP和血红蛋白这两种标准蛋白质样品对接口性能进行评估。实验结果表明,该方法具有较高的灵敏度和分离度且解决了电泳过程中H2O2电解产生气泡等问题。

图1 旋转检测池的示意图和照片[49]Fig. 1 Schematic diagram and photographs of the rotary detection cell[49] a and b: top and bottom views; c and d: photos in close and general views. 1. step motor and lead screw; 2. detection cell body; 3. Pt cathode; 4. slit; 5. quartz bottom; 6. photo multiplier tube (PMT); 7. separation capillary; 8. offset between capillary and the slit window.
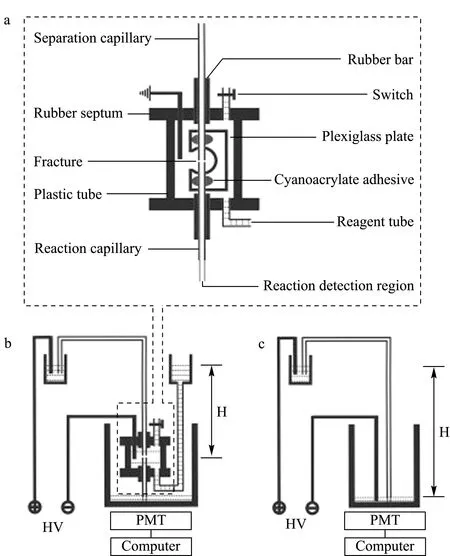
图2 (a)在柱断口、(b)在柱断口/柱端反应接口和(c)传统的柱端CE-CL接口示意图[50]Fig. 2 Schematic diagrams of (a) the on-column fractureassembly, (b) the on-column fracture/end-column reaction interface and (c) the conventional end-column capillary electrophoresis-chemiluminescence (CE-CL) interface[50] H: height; HV: high voltage.
Xu等[50]提出一种新的接口方式,可以分别完成CE分离和CL检测条件的优化。他们将在柱裂缝浸于阴极缓冲液池内完成电路连接并用作CL试剂加入口,再将毛细管末端插入柱端蓄池内完成CL反应及检测,其结构见图2。这种设计结合了离柱式和柱端式接口的优点,并且消除了各自的局限性。通过吖啶酯-luminol反应体系对该接口适用性进行评估,结果发现该方法较之前的接口方式表现出更好的分析性能。Xie等[51]设计了一种新型的固定化酶检测接口,通过利用共价键合作用将HRP固定于醛基纸膜上,在自行设计的固定化酶发光流通池中催化luminol-H2O2-溴酚蓝CL体系。该实验将含有巯基的β-环糊精修饰的纳米金作为固定相涂于毛细管内壁中制作开管毛细管电色谱柱,并采用三乙胺催化生物胺与N-(4-氨基丁基)-N-乙基异鲁米诺(ABEI)的衍生反应,通过开管毛细管电色谱-CL联用技术对人体唾液及尿液样品进行了分析检测。
3 应用
CE与CL联用系统自20世纪90年代出现以来就一直受到广泛关注,并应用于药物分析[16,34,52-80]、蛋白质分析[81-84]、细胞分析[85-90]以及基于纳米材料的分析[91-101]等领域。
3.1 药物分析
液相色谱法在药物检验中应用广泛,但系统操作和维护较为复杂,并消耗大量的有机溶剂。CE-CL联用方法由于其费用低、操作简单、分析时间短等特点成了药物检测的一种新方法,其原理主要基于药物对CL反应的增强或抑制效应。近年来,CE-CL检测用于药物分析的部分研究如表1所示[16,34,52-79]。
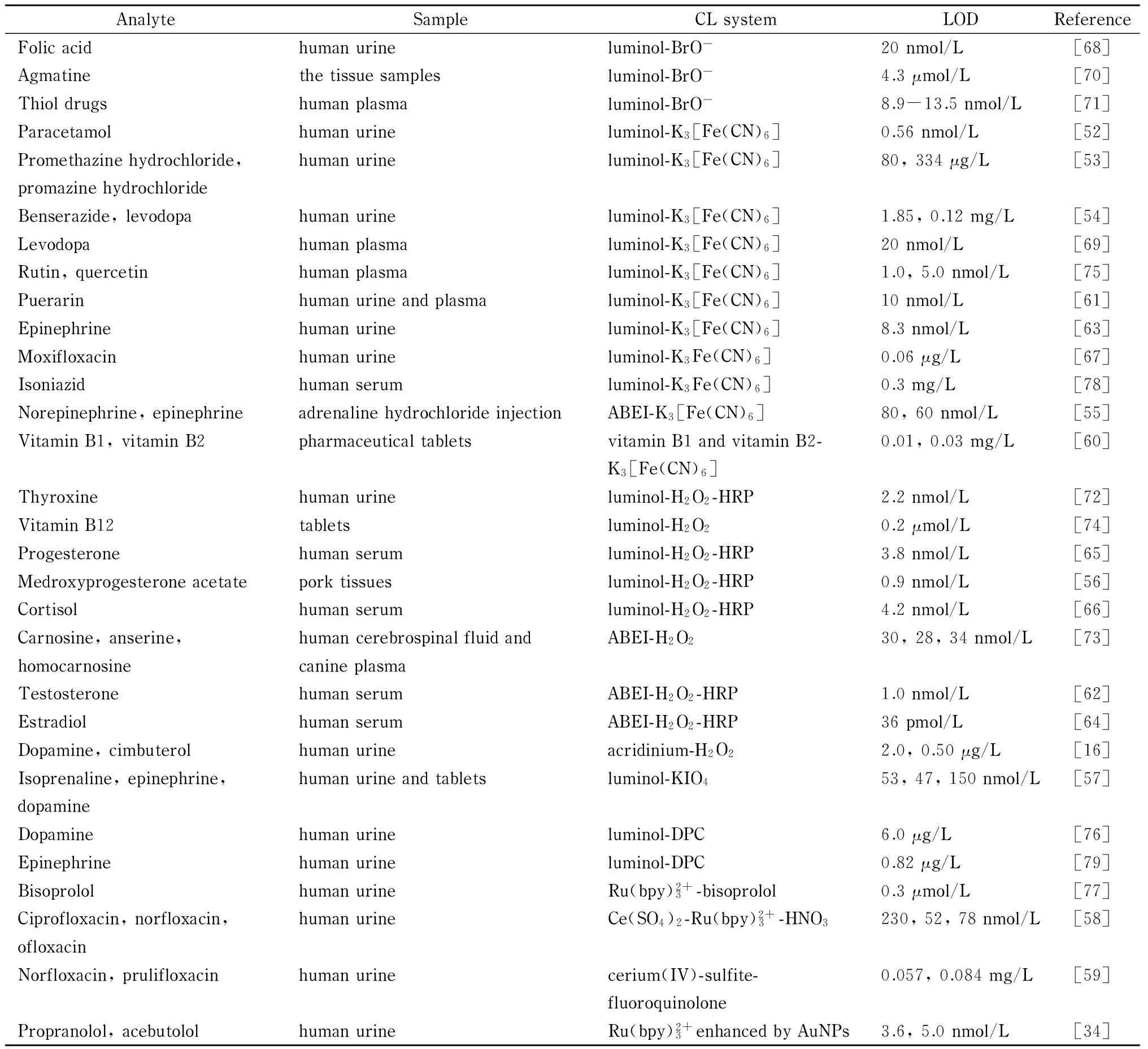
表1 近年来CE-CL检测在药物分析中的应用Table 1 Applications of CE-CL in drug analysis in recent years
CN: cyanide; ABEI:N-(4-aminobutyl)-N-ethylisoluminol; HRP: horseradish peroxidase; DPC: diperiodatocuprate (Ⅲ); bpy: bipyridine; AuNPs: Au nanoparticles.
Zhao等[52,62,64-66,68-73]对CE-CL联用方法在药物方面的应用做了一系列深入研究,他们提出了一种简单快速的方法检测苹果汁以及人体尿液中的叶酸[68]。该方法基于叶酸对luminol-BrO-反应体系的增强效应。该方法在20 min之内完成对叶酸的测定,检出限达到2.0×10-8mol/L,峰面积和迁移时间的相对标准偏差分别为1.5%和1.1%。他们还通过这种简单快速的方法检测药物制剂及生物流体中的左旋多巴[69]。该方法主要基于左旋多巴对luminol-K3[Fe(CN)6]反应体系的增强效应。在最佳实验条件下,线性范围为5.0×10-8~2.5×10-6mol/L,检出限为2.0×10-8mol/L。
Zhu等[74]基于维生素B12对luminol-H2O2反应体系的催化活性建立了CE-CL检测方法。在对药品中维生素B12的加标回收试验中,回收率达到了97%~106%。
Wang等[75]采用CE-CL检测方法测定药品和人血浆中的黄酮类化合物(芦丁和槲皮素)的含量。芦丁和槲皮素可以增强luminol-K3[Fe(CN)6]反应体系中的CL信号,两种物质的检出限分别为1和5 nmol/L。该方法还可以对复杂样品中黄酮类化合物进行定量分析。
过渡金属在高氧化态时不稳定,Wang等[76]将二过碘酸合铜(Ⅲ)钾(K5[Cu(HIO6)2], DPC)用于CE-CL系统中检测多巴胺。该方法主要基于多巴胺增强luminol-DPC体系的CL信号。多巴胺的检出限达6.0×10-6g/L,较于之前用其他方法检测多巴胺的报道,该方法具有更高的灵敏度。DPC可以增强luminol-H2O2的CL反应并产生强烈的CL信号。基于此,Fu等[80]建立了一种CE-CL快速检测体系对luminol和ABEI进行同时检测。实验结果表明该方法可以用于检测复杂的生物样品,在生物医学和药物分析等领域具有良好的应用前景。

3.2 蛋白质分析
蛋白质是生命的物质基础,与各种形式的生命活动紧密联系在一起。因此对蛋白质进行分离和分析在生命科学方面具有重要意义。
Zhou等[81]采用CE-CL联用方法研究血红素(hemoglobin,Hb)和H2O2的反应过程。该方法基于Hb和Fe3+对H2O2-CL反应体系的增强作用对Hb和Fe3+进行检测。Hb和H2O2反应会产生荧光产物导致Hb释放Fe3+。在该反应过程中,有一个中间产物可以显著增强luminol-H2O2反应体系的CL信号。Fe3+、Hb及该中间产物的最大CL信号比大约为1∶10∶60。
为了解决蛋白质非特异性吸附问题,Liu等[82]设计了一种动态涂层CE-CL检测系统。聚乙烯吡咯烷酮具有易溶于水且黏度低于其他高分子溶液的特点,可以轻松注入毛细管且涂覆于毛细管内壁。实验结果表明,注入聚乙烯吡咯烷酮后可以很大程度抑制蛋白质吸附并降低电渗流。涂层的毛细管至少可以连续30次注入蛋白质样品并具有良好的重复性。该方法成功用于检测溶血病患者血清中Hb的浓度,检出限为2.0 mg/L。
Zhang等[83]采用HRP标记乙型肝炎表面抗原(hepatitis B surface antigen,HBsAg)形成HBsAg*,引入对碘苯酚增强CL反应。该方法分别采用竞争免疫和非竞争免疫形式检测人体血清中HBsAg与乙型肝炎表面抗体(HBsAb)。游离的HBsAg*和结合的HBsAg*-HBsAb在6 min内通过CE分离,HBsAg和HBsAb的检出限分别为0.4 pmol/L和1 IU/L。该实验结果与酶联免疫吸附测定的结果相一致,证明CE-CL免疫测定法在临床诊断上具有可行性。Liu等[84]同样采用CE-CL免疫测定法分析肿瘤标记物甲胎蛋白。甲胎蛋白抗原与过量HRP标记的抗体Ab*反应,游离的Ab*与生成的Ab*-Ag通过CE在4 min内完成分离,检出限为0.85 μg/L。
3.3 细胞分析
分析单细胞内组分及其行为特征有助于了解基本的细胞功能,而微流控芯片的微米尺寸通道适合单细胞的引入、反应、分离及检测。因此,CE-CL、MCE-CL分离分析方法适用于单细胞分析。
本课题组[85]提出采用CE-CL检测方法对单个红细胞内Hb的含量进行测定,并对细胞注射流程进行了改善。通过该方法检测了人血液样本中Hb的含量,线性范围为1.7~6.8 mg/L,检出限为0.17 mg/L(乘以进样体积之后约为2.2 pg)。通过对26个人体红细胞样本统计表明,单个红细胞中Hb的平均含量为23.6 pg。
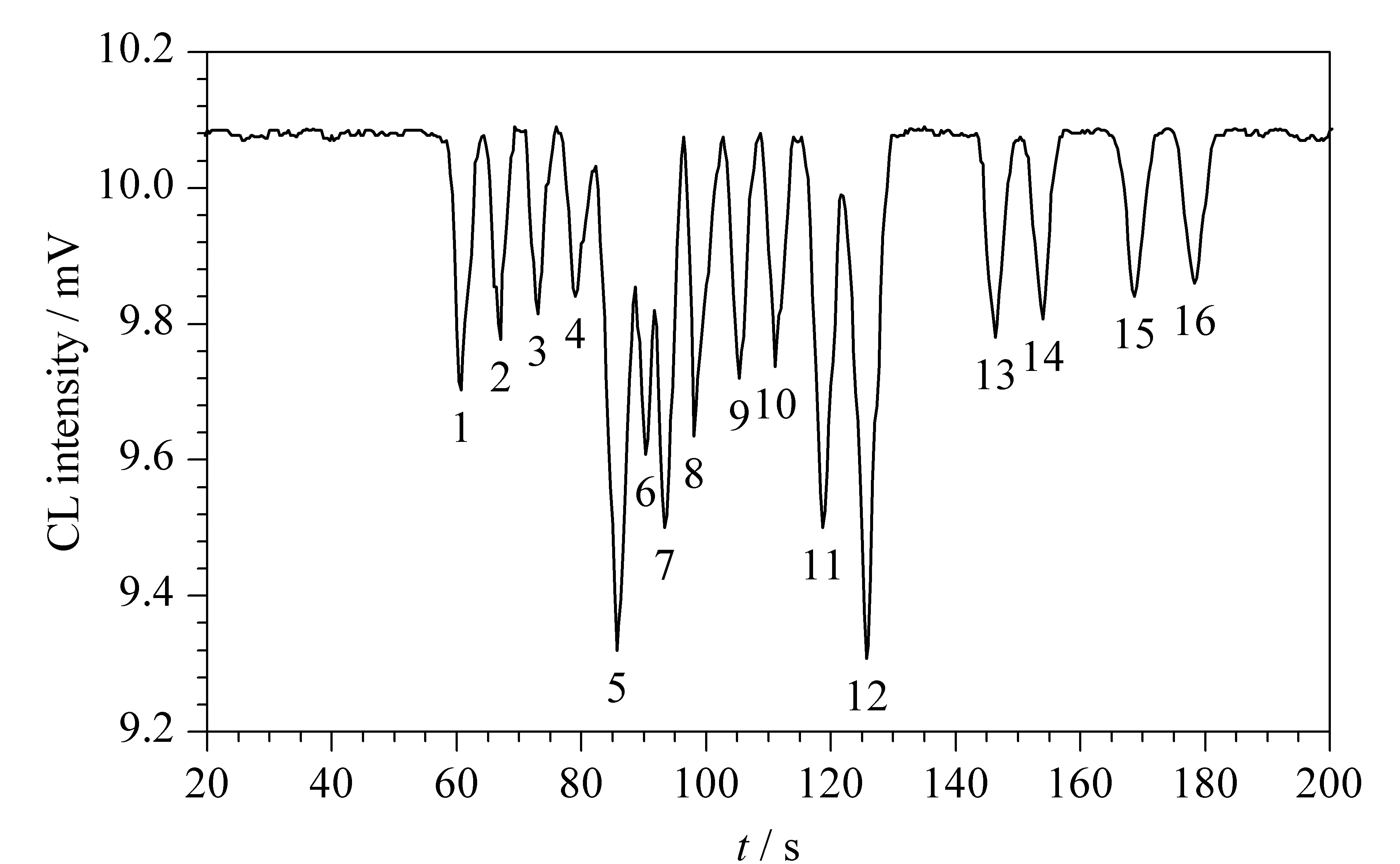
图3 20种氨基酸的电泳图谱[87]Fig. 3 Electropherogram of the 20 amino acids[87] Peaks: 1. Arg; 2. Lys; 3. Ala; 4. Pro+Val; 5. Leu+Ile; 6. Phe+His; 7. Tyr; 8. Gln+Met; 9. Ser; 10. Asn; 11. Thr; 12. Cys; 13. Gly; 14. Trp; 15. Glu; 16. Asp.
Zhao等[86]采用MCE-CL方法对红细胞内谷胱甘肽的含量进行测定。首先采用重氮luminol标记细胞内的谷胱甘肽,然后单细胞进样,在线加电压解离。样品通过MCE分离后,与次溴酸钠发生氧化反应产生CL。检出限达5×10-20mol,线性范围为2×10-18~9.0×10-17mol。相较于之前报道的激光诱导荧光方法检测单细胞内谷胱甘肽含量,该方法更简单,且灵敏度高出100倍左右。该课题组还提出了一种高灵敏度的方法测定单个人体血红细胞中氨基酸的含量[87],图3是采用该方法分析20种氨基酸的电泳图谱。发现在luminol-NaBrO-QD(quantum dots)体系中具有高效的能量共振转移,并且受到生物胺类、生物硫醇、氨基酸及有机酸等的有效抑制。生物胺类的检出限可达10-9mol/L,生物硫醇、有机酸、雌激素及天然氨基酸的检出限为10-8mol/L。结果表明,该方法比之前报道的MCE-CL检测法、电化学法等的灵敏性高10~1 000倍。
Zhao等[88]采用同样的方法,检测了大鼠肝细胞中抗坏血酸和氨基酸(色氨酸、甘氨酸和丙氨酸)的含量。这些物质在130 s内完成分离,在单个大鼠肝细胞中的含量分别为38.3、5.15、3.78和3.84 fmol。同样,Ye等[89]对大鼠纤维肉瘤细胞中牛磺酸和氨基酸(丙氨酸、甘氨酸、色氨酸、谷氨酸和天冬氨酸)的含量也进行了检测。除此之外,他们还对血红细胞中的巯基化合物(半胱氨酸、谷胱甘肽和Hb)含量进行了测定[90],检出限分别为7、32和69 amol。对正常人及癌症患者的血红细胞分析结果表明,癌症患者血红细胞内半胱氨酸、谷胱甘肽的含量均高于正常人。因此可以通过该方法量化血红细胞内的半胱氨酸和谷胱甘肽,为癌症临床诊断提供一种方法。
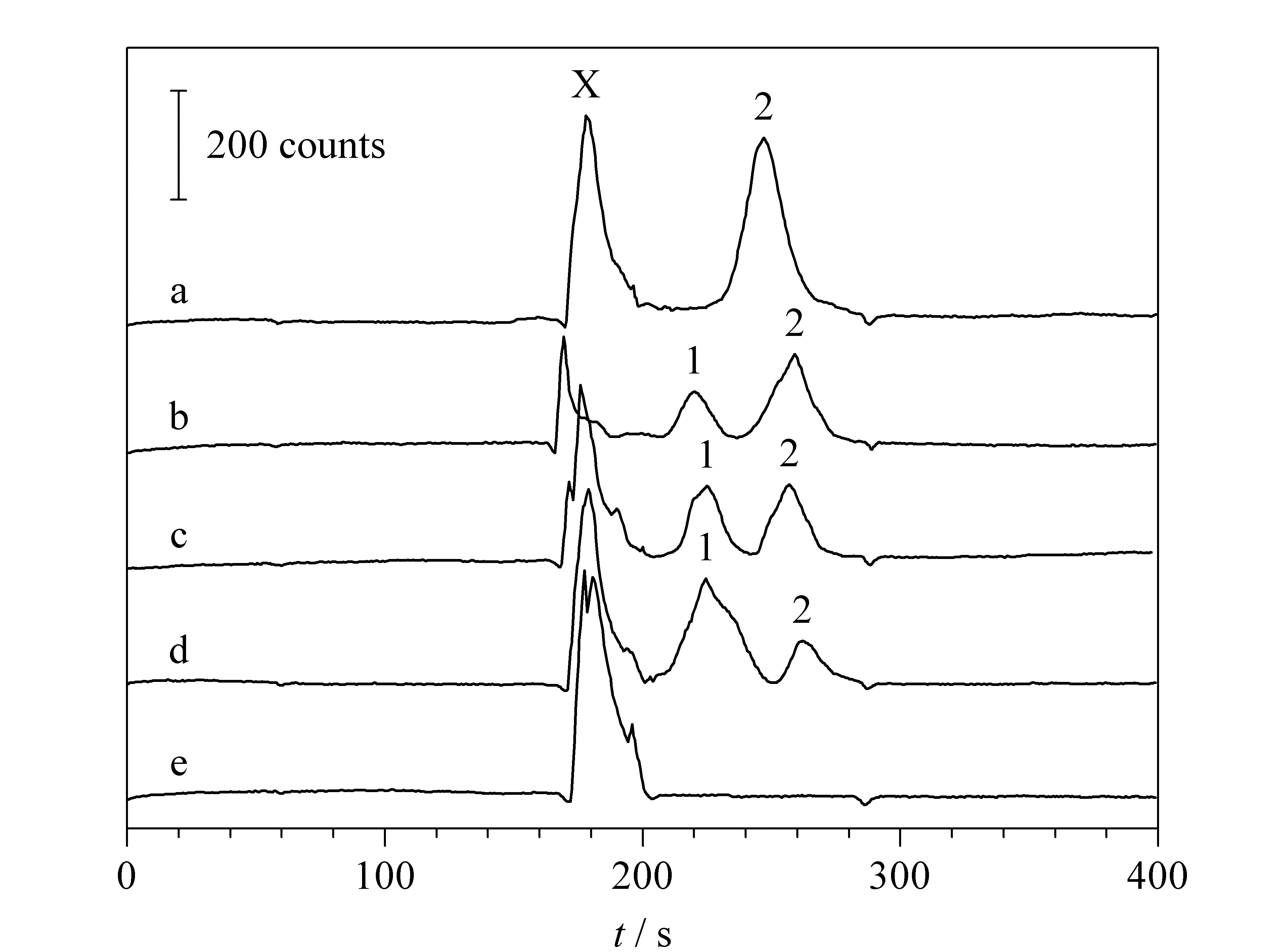
图4 Ab*与免疫复合物的电泳图谱[94]Fig. 4 Electropherograms of Ab*and the immunocomplex[94] Lines a, b, c, and d correspond to 0, 0.1, 0.5, and 3.0 μg/mL human IgG, respectively; Line e: blank. Peaks: 1. Immunocomplex; 2. free Ab*; X. unknown compounds.
3.4 基于纳米材料的分析
纳米材料如AuNPs[91-96]、QDs[97-99]、氧化石墨烯[100](grapheme oxide, GO)以及纳米介孔二氧化硅(mesoporous silica nanoparticles, MSN)[101]等已经用于高灵敏度CE-CL的检测中。
Liu等[94]提出基于AuNPs的CE-CL免疫实验方法检测人血清中免疫球蛋白G。该方法采用对luminol-H2O2反应体系有催化作用的AuNPs来标记蛋白质。AuNPs和抗体Ab结合形成标记抗体Ab*, Ab*随后与抗原Ag反应生成Ab*-Ag,图4为Ab*与免疫复合物的电泳图谱。过量的Ab*及Ab*-Ag在5 min内完成分离和检测,检出限为1.14×10-3mg/L。基于此,Liu等[95]随后提出了基于核酸适配体(aptamer,Apt)的CE-CL方法对人免疫球蛋白E进行检测,检出限达到7.6 fmol/L,线性范围为0.025~250 pmol/L。实验结果表明该方法可用于生物样品中蛋白质的高灵敏度检测。
Jiang等[96]用AuNPs作为载体、luminol-H2O2为发光体系的非竞争性免疫分析方法检测人体血清中的癌胚胎抗原(carcinoembryonic antigen, CEA)。AuNPs结合HRP共同标记癌胚胎抗体Ab*并与CEA共同孵育。随后通过CE-CL系统对CEA-Ab*-AuNPs和过量的Ab*-AuNPs进行分离和定量分析。通过对碘苯酚增强以及AuNPs放大luminol-H2O2-HRP系统的CL信号使得CL检测具有高灵敏度,检出限达到0.034 μg/L。
QD具有很好的光电学性能,被用于高灵敏CE-CL分析技术中。Zhou等[98]提出一种测定CEA的新方法,主要采用QD修饰Apt探针。DNAA与DNAB为一对互补的核酸序列。HRP-DNAA首先与QD-DNAB形成HRP-DNAA-B-QD探针。当CEA存在时,DNAA中Apt与CEA特异性结合形成CEA/HRP-DNAA-B-QD复合物。HRP-DNAA-B-QD探针和CEA/HRP-DNAA-B-QD复合物通过CE分离后,CEA/HRP-DNAA-B-QD复合物在无任何干扰下催化CL,通过CL强度可以估算该复合物浓度。该方法中,QD的引入解决了游离HRP-DNAA-B-QD探针干扰的问题,为分离检测生物分子提供了新的思路。Zhang等[99]采用CE-CL检测方法同时检测5-羟吲哚乙酸和5-羟色胺。CdTe(cadmium telluride) QDs和HRP共同催化luminol-H2O2CL反应,增强CL信号强度,而微量的5-羟吲哚乙酸和5-羟色胺可以抑制CL。目前,该方法已经成功对人体尿液中5-羟吲哚乙酸和5-羟色胺进行检测,检出限分别为7.0和6.0 nmol/L。
Zhou等[100]以GO为载体、luminol-H2O2为发光体系对CEA进行检测(见图5)。当无CEA存在时,HRP-Apt与GO混合产生能量共振转移从而淬灭CL;当CEA存在时,HRP-Apt与CEA特异性结合形成HRP-Apt-CEA复合物,从GO上分离。随后该复合物催化产生CL而没有检测到任何能量转移。该方法通过HRP-Apt吸附在GO上解决了游离HRP-Apt的干扰问题,同时GO对CL的影响可以通过CE解决。实验结果表明,CL强度与CEA的浓度呈线性关系,线性范围为0.065 4~6.54 μg/L,检出限为4.8 ng/L。该方法具有较高的特异性,为生物大分子的分离和测定提供了新方法,在生化分析中有很好的应用潜力。
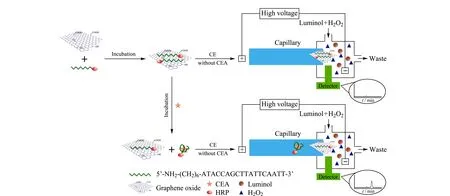
图5 基于Apt/GO CE-CL检测方法检测CEA的示意图[100]Fig. 5 Schematic depiction of detection of carcinoembryonic antigen (CEA) based on aptamer/grapheme oxide (Apt/GO) by CE-CL[100]
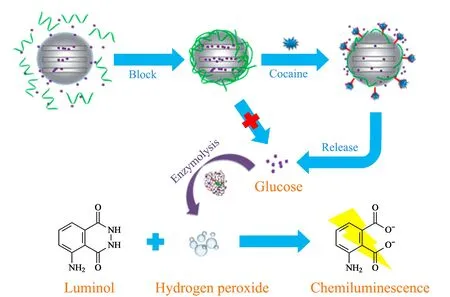
图6 纳米介孔二氧化硅可控释放系统与CL检测技术联用系统检测可卡因的示意图[101]Fig. 6 Schematic illustration of the mesoporous silicananoparticles (MSN) based controlled release system coupled with luminol/H2O2CL system for cocaine determination[101]
纳米介孔二氧化硅具有较高的比表面积,较高的孔隙率,孔径分布均匀且小,表面富含活性羟基,在孔内和孔外实现各种官能团化以及良好的生物兼容性等特点,在药物可控释放方面具有广泛的应用前景。Chen等[101]首次将纳米介孔二氧化硅可控释放系统与CL检测技术联用,建立了一个灵敏的生物传感器并成功应用于人尿液中可卡因的检测(见图6)。在该方法中,首先将MSN载体与葡萄糖溶液混合,MSN壁与葡糖糖分子之间的强烈作用力使大量葡萄糖分子负载于介孔孔道内;之后带正电荷的MSN与带负电荷的寡核苷酸(可卡因Apt)反应关闭了MSN孔,形成“孔帽”。当可卡因存在时,可卡因与其Apt间的特异性识别,导致“孔帽”移走,孔道内的葡萄糖释放出来。释放出的葡萄糖在葡萄糖氧化酶的存在下与氧气反应产生葡萄糖酸与H2O2,后者可以增强luminol的CL,检出限达到1.43 μmol/L。该方法可以通过设计不同的分子探针实现对不同药物的检测。
3.5 其他
CE-CL联用检测方法还应用于金属离子[102,103]、氨基酸[88,104,105]、糖类[106]、有机小分子[107-110]等领域的分析。
CE-CL检测方法最初多用于对金属离子的分离分析,主要基于金属离子对luminol-H2O2反应体系的催化作用。Yang等[102]采用luminol-H2O2反应体系测定水中污染金属离子Cr(Ⅲ)、Cr(Ⅵ)。他们将Cr(Ⅵ)转化为Cr(Ⅲ),实现分离检测,Cr(Ⅲ)和Cr(Ⅵ)的检出限分别为6×10-13和8×10-12mol/L。
基于Cu(Ⅱ)-氨基酸复合物在碱性条件下对luminol-H2O2反应体系的增强作用,Lin等[104]采用加压毛细管电色谱与CL联用方法对L-组氨酸、L-苏氨酸和L-酪氨酸进行分离分析。结果表明,L-组氨酸、L-苏氨酸和L-酪氨酸的检出限分别为6.4×10-7、8.4×10-7和3.0×10-7mol/L。Li等[105]在柱前通过ABEI将生物胺衍生化,并在柱后采用高灵敏度的luminol-H2O2-DPC反应体系进行检测。通过该方法得到的背景信号很低且CL信号较强,灵敏度较其他方法有进一步提高。实验结果表明,甘氨酸、脯氨酸和苯基丙氨酸的检出限分别为3.0×10-8、2.3×10-7和2.1×10-7mol/L。
Tsukagoshi等[106]通过单糖、二糖的二醇基与碘代苯硼酸之间的相互反应对单糖、二糖进行分子识别。其中,碘代苯硼酸可以增强luminol-H2O2-HRP反应体系的CL信号,但其与糖类反应后会使CL信号降低从而成功实现识别。相较于传统的方法,该方法对于糖类分子识别所需要的样品量更少,分析更加快速,并且简单易操作。
Shu等[109]基于Hb的催化性能,采用CE与Hb-luminol-H2O2的CL体系联用方法对酚类物质进行分离和检测。高浓度的NH4OH可以防止Hb的聚集,得到稳定的基线信号。该方法用于对邻仲丁基苯酚、苯酚、2,4-二氯苯酚、邻甲苯酚及间甲苯酚的测定,检出限在4.8×10-8~7.1×10-8mol/L之间。Lin等[110]基于苯二酚的3种同分异构体可以抑制luminol-K3[Fe(CN)6]反应体系的CL,对尿液中苯二酚的3种同分异构体(邻苯二酚、间苯二酚和对苯二酚)进行测定,检出限分别为2.8×10-8、3.2×10-8和3.7×10-8mol/L。
4 总结与展望
CL检测系统因其无需外加光源、不存在荧光分析中瑞利散射和拉曼散射及溶剂荧光杂质产生的背景信号,具有较高的信噪比,可获得与激光诱导荧光检测相媲美的灵敏度。CE和MCE具有分析速度快,样品用量少和分离效率高等特点。CE和CL的联用兼具高分离效率与高灵敏度的特点,已发展成为一种高效的分离检测方法。多种可联用CE的发光体系及可用的接口设计模式使这一方法得到较快的发展,在生物制药、环境分析和临床医学等领域获得了广泛的应用。
然而,CE-CL联用方法的重现性仍然不太理想。这主要是由于该方法的重现性除了与电泳条件相关外,还受化学发光反应类型、反应物混合模式和反应物流速的稳定性等因素的影响。另外,相对紫外可见吸收法和激光诱导荧光法,该方法的线性范围较窄(约两个数量级)。此外,可以直接参与CL反应的待测物种类不多,相关标记技术也不是很成熟,因此如何扩大CE-CL中分析对象的范围成了现实的问题。
目前,CE-CL联用方法尚需要在以下几个方面继续开展研究:(1)设计更简洁的CE-CL联用仪器接口,进一步提高系统的重现性、便捷性、选择性和灵敏度。另外,CE-CL联用方法可与其他检测方法如质谱联用从而获得更好的性能;(2)将新型的CL试剂和纳米材料如AuNPs、QD和荧光高分子纳米粒子等应用于CE-CL联用系统,进一步拓宽线性范围,提高灵敏度,扩展其应用范围;(3)新方法(如化学发光共振能量转移方法)的进一步应用将为CE-CL联用方法提供更高的灵敏度以及进一步满足在复杂体系中同时分离检测多种物质的需求;(4)可以尝试应用毛细管阵列电泳及芯片毛细管阵列电泳与CL检测联用实现高通量样品分析。
[1] Dumke J C, Nussbaum M A. Anal Chem, 2007, 79: 1262
[2] Lee T T, Yeung E S. J Chromatogr, 1992, 595: 319
[3] Evangelista R A, Liu M, Chen F A. Anal Chem, 1995, 67: 2239
[4] Zhao W F, Chen Q D, Wu R G, et al. Electrophoresis, 2011, 32: 3025
[5] Gavin P F, Ewing A G. J Am Chem Soc, 1996, 118: 8932
[6] Guo L H, Yang H H, Qiu B. Anal Chem, 2009, 81: 9578
[7] Li J J, Kelly J F, Chernushevich I, et al. Anal Chem, 2000, 72: 599
[8] Liu Y X, Huang X Y, Ren J C. Electrophoresis, 2016, 37: 2
[9] Xu X D, Zhang H Y, Shi H M, et al. Anal Biochem, 2012, 427: 10
[10] Liu B F, Ozaki M, Utsumi Y, et al. Anal Chem, 2003, 75: 36
[11] Liu Y M, Liu Y Y, Zhou M, et al. J Chromatogr A, 2014, 1340: 128
[12] Xie H Y, Wang Z R, Fu Z F. J Pharm Anal, 2014, 4(6): 412
[13] Albrecht H O. Chemical, 1928, 136: 321
[14] Hashimoto M, Tsukagoshi K, Nakajima R, et al. J Chromatogr A, 2000, 867: 271
[15] Tsukagoshi K, Obata Y, Nakajima R. J Chromatogr A, 2002, 971: 255
[16] Wang Z R, Yue H, Wang Y H, et al. Electrophoresis, 2014, 35: 1000
[17] Yu H, Xu L, You T Y. Luminescence, 2013, 28: 217
[18] Xu L, Li L B, Huang J S, et al. Talanta, 2014, 118: 1
[19] Tsukagoshi K, Okuzono N, Nakajima R. J Chromatogr A, 2002, 958: 283
[20] Lara F J, Airado-Rodríguez D, Moreno-González D, et al. Anal Chim Acta, 2016, 913: 22
[21] Zhang X F, Xuan Y L, Sun A M, et al. Luminescence, 2009, 24: 243
[22] Han S Q, Wang H L. J Chromatogr B, 2010, 878: 2901
[23] Han S Q, Wu K G. J Chin Chem Soc, 2015, 62: 73
[24] Zhu J K, Shu L, Wu M, et al. Talanta, 2012, 93: 428
[25] Mu X M, Li S T, Lu X, et al. Electrophoresis, 2014, 35: 962
[26] Tsuge K, Tanaka T, Noda K, et al. J Liq Chromatogr Related Technol, 2012, 35: 1091
[27] Zhu J K, Shu L, Zhang F, et al. Luminescence, 2012, 27: 482
[28] Guo L H, Qiu B, Chen M X, et al. Electrophoresis, 2009, 30: 1355
[29] García-Campaa A M, Lara F J, Gámiz-Gracia L, et al. TrAC-Trends Anal Chem, 2009, 28: 973
[30] Barnett N W, Hindson B J, Lewis S W. Analyst, 2000, 125: 91
[31] Xu X Q, Wang J P, Wen F Y, et al. Anal Methods, 2015, 7: 976
[32] Shi G Y, Li J, Yin Y C, et al. Luminescence, 2013, 28: 468
[33] Hassanzadeh J, Amjadi M. Luminescence, 2015, 30: 439
[34] Duan H B, Cao J T, Wang H, et al. RSC Adv, 2016, 6: 45533
[35] Su M, Wei W, Liu S Q. Anal Chim Acta, 2011, 704: 16
[36] Guo L H, Fu F F, Chen G N. Anal Bioanal Chem, 2011, 399: 3323
[37] Ren J C, Huang X Y. Anal Chem, 2001, 73: 2663
[38] Yang W P, Zhang Z J, Deng W. J Chromatogr A, 2003, 1014: 203
[39] Hara T, Okamura S, Kato J, et al. Anal Sci, 1991, 7: 261
[40] Dadoo R, Colon L A, Zare R N, et al. J High Resol Chromatogr, 1992, 15: 133
[41] Liao S Y, Chao Y C, Whang C W. J High Resol Chromatogr, 1995, 18: 667
[42] Liao S Y, Whang C W. J Chromatogr A, 1996, 736: 247
[43] Jiang H L, He Y Z, Zhao H Z, et al. Anal Chim Acta, 2004, 512: 111
[44] Lee Y T, Whang C W. J Chromatogr A, 1997, 771: 379
[45] Liu Y M, Cheng J K. J Chromatogr A, 2003, 1003: 211
[46] Lin Z A, Sun X B, Lin Y, et al. Analyst, 2013, 138: 2269
[47] Dadoo R, Seto A G, Colón L A, et al. Anal Chem, 1994, 66: 303
[48] Tsukagoshi K, Nakahama K, Nakajima R. Anal Chem, 2004, 76: 4410
[49] Wang J H, Li L M, Huang W H, et al. Anal Chem, 2010, 82: 5380
[50] Xu Q F, Ji X H, Li H G, et al. J Chromatogr A, 2010, 1217: 5628
[51] Xie H Y, Wang Z R, Kong W J, et al. Analyst, 2013, 138: 1107
[52] Zhao S L, Bai W L, Yuan H Y, et al. Anal Chim Acta, 2006, 559: 195
[53] Lara F J, García-Campaa A M, Gámiz-Gracia L, et al. Electrophoresis, 2006, 27: 2348
[54] He W W, Zhou X W, Lu J Q. J Chromatogr A, 2006, 1131: 289
[55] Lin Z A, Wu X P, Lin X C, et al. J Chromatogr A, 2007, 1170: 118
[56] Peng C F, Huo T M, Liu L Q, et al. Electrophoresis, 2007, 28: 970
[57] Liu Y M, Wang C Q, Mu H B, et al. Electrophoresis, 2007, 28: 1937
[58] Liu Y M, Jia Y X, Tian W. J Sep Sci, 2008, 31: 3765
[59] Yang Z J, Wang X L, Qin W D, et al. Anal Chim Acta, 2008, 623: 231
[60] Bai J, Hun X, Liu Q, et al. Microchim Acta, 2008, 160: 165
[61] Han S Q. J Chromatogr B, 2009, 877: 1591
[62] Huang Y, Shi M, Zhao S L, et al. Electrophoresis, 2011, 32: 3196
[63] Su Y Y, Chen C, Hou X L, et al. Anal Methods, 2011, 3: 2893
[64] Zhao S L, Liu J W, Huang Y, et al. Chem Commun, 2012, 48: 699
[65] Ye F G, Liu J W, Huang Y, et al. J Chromatogr B, 2013, 936: 74
[66] Ye F G, Yang T Z, Huang Y, et al. Anal Methods, 2013, 5: 5657
[67] Suh Y S, Kamruzzaman M, Alam A M, et al. Luminescence, 2014, 29: 248
[68] Zhao S L, Yuan H Y, Xie C, et al. J Chromatogr A, 2006, 1107: 290
[69] Zhao S L, Bai W L, Wang B, et al. Talanta, 2007, 73: 142
[70] Zhao S L, Xie C, Lu X, et al. J Chromatogr B, 2006, 832: 52
[71] Huang Y, Zhao S L, Shi M, et al. J Pharmaceut Biomed Anal, 2011, 55: 889
[72] Huang Y, Zhao S L, Shi M, et al. Anal Biochem, 2010, 399(1): 72
[73] Zhao S L, Huang Y, Shi M, et al. Anal Biochem, 2009, 393(1): 105
[74] Zhu J K, Shu L, Wu M, et al. Journal of East China Normal University (Natural Science), 2013, 32(5): 96
朱金坤, 舒露, 吴敏, 等. 华东师范大学学报(自然科学版), 2013, 32(5): 96
[75] Wang J, Han S Q. Chromatographia, 2013, 76: 715
[76] Wang L, Liu Y, Xie H Y, et al. Electrophoresis, 2012, 33: 1589
[77] Wang J W, Zhang X J, Pi F F, et al. Electrochim Acta, 2009, 54: 2379
[78] Liu Y, Fu Z F, Wang L. Luminescence, 2011, 26: 397
[79] Li T, Wang Z R, Xie H Y, et al. J Chromatogr B, 2012, 911: 1
[80] Fu Z F, Li Z Y, Xie H Y, et al. Electrophoresis, 2010, 31: 3342
[81] Zhou S L, Wang J H, Huang W H, et al. J Chromatogr B, 2007, 850: 343
[82] Liu H Y, Han N, Zhang L Y, et al. Anal Chim Acta, 2010, 680: 48
[83] Zhang Y X, Zhang Z J, Yang F. J Chromatogr B, 2007, 857: 100
[84] Liu Y M, Mu H B, Zheng Y L, et al. J Chromatogr B, 2007, 855: 280
[85] Zhi Q, Xie C, Huang X Y, et al. Anal Chim Acta, 2007, 583: 217
[86] Zhao S L, Li X T, Liu Y M. Anal Chem, 2009, 81: 3873
[87] Zhao S L, Huang Y, Shi M, et al. Anal Chem, 2010, 82: 2036
[88] Zhao S L, Huang Y, Liu Y M. J Chromatogr A, 2009, 1216(39): 6746
[89] Ye F G, Huang Y, Xu Q, et al. Electrophoresis, 2010, 31: 1630
[90] Zhao S L, Huang Y, Ye F G, et al. J Chromatogr A, 2010, 1217: 5732
[91] Zhao S L, Niu T X, Song Y R, et al. Electrophoresis, 2009, 30: 1059
[92] Zhao S L, Lan X H, Liu Y M. Electrophoresis, 2009, 30(15): 2676
[93] Liu Y M, Mei L, Liu Y Y, et al. Electrophoresis, 2014, 35: 972
[94] Liu Y M, Mei L, Liu L J, et al. Anal Chem, 2011, 83: 1137
[95] Liu Y M, Cao J T, Liu Y Y, et al. Electrophoresis, 2015, 36: 2413
[96] Jiang J, Zhao S L, Huang Y, et al. J Chromatogr A, 2013, 1282: 161
[97] Zhao Y S, Zhao S L, Huang J M, et al. Talanta, 2011, 85: 2650
[98] Zhou Z M, Feng Z, Zhou J, et al. Sens Actuators B, 2015, 210: 158
[99] Zhang L L, Zhao Y S, Huang J M, et al. J Chromatogr B, 2014, 967: 190
[100] Zhou Z M, Feng Z, Zhou J, et al. Biosens Bioelectron, 2015, 64: 493
[101] Chen Z H, Tan Y, Xu K F, et al. Biosens Bioelectron, 2016, 75: 8
[102] Yang W P, Zhang Z J, Deng W. Anal Chim Acta, 2003, 485: 169
[103] Yang W P, Zhang Z J, Deng W. J Chromatogr A, 2003, 1014: 203
[104] Lin Z A, Xie Z H. J Sep Sci, 2008, 31: 2852
[105] Li T, Xie H Y, Fu Z F. Anal Chim Acta, 2012, 719: 82
[106] Tsukagoshi K, Matsumoto K, Ueno F, et al. J Chromatogr A, 2006, 1123: 106
[107] Ji X H, He Z K, Pang D W. Electrophoresis, 2007, 28: 3260
[108] Hu H M, Yin X F, Wang X Z, et al. J Sep Sci, 2013, 36: 713
[109] Shu L, Zhu J K, Wang Q J, et al. Luminescence, 2014, 29: 579
[110] Lin Z A, Sun X B, Hu W L, et al. Electrophoresis, 2014, 35: 993
Recent advances in capillary electrophoresis coupledwith chemiluminescence detection
YI Fang, HUANG Xiangyi*, REN Jicun*
(SchoolofChemistryandChemicalEngineering,StateKeyLaboratoryofMetalMatrixComposites,ShanghaiJiaotongUniversity,Shanghai200240,China)
Capillary electrophoresis (CE) is considered as a powerful method in many fields, such as biopharmaceuticals, environmental, food and public security analysis, owing to its high separation efficiency. However, the injection of small sample volumes and the short optical length lead to limited sensitivity. So it is necessary to couple with high sensitivity detector to realize the low concentration sample analysis. Chemiluminescence (CL) detection is characterized by providing low background with excellent sensitivity. By coupling with CL, both the high separation efficiency of CE and the high sensitivity of CL can be achieved at the same time. So far, this method has been widely applied to chemical analysis, drug screening, and environment monitoring. In this review, we briefly introduce some developments for CE-CL systems, and then put the emphasis on the applications in the past decades. Moreover, we discuss the perspectives of CE-CL system.
capillary electrophoresis (CE); chemiluminescence (CL) detection; review
10.3724/SP.J.1123.2016.08043
2016-08-31
国家自然科学基金项目(21075081,21135004);上海市自然科学基金项目(14ZR1423400).
Foundation item: National Natural Science Foundation of China (Nos. 21075081, 21135004); Shanghai Natural Science Foundation (No. 14ZR1423400).
O658
:A
:1000-8713(2017)01-0110-11
*通讯联系人.E-mail:huangxy@sjtu.edu.cn(黄香宜);E-mail:jicunren@sjtu.edu.cn(任吉存).
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
云南化工(2020年11期)2021-01-14 00:50:54
装备制造技术(2019年12期)2019-12-25 03:07:10
上海建材(2017年4期)2017-04-06 07:32:03
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
杂草学报(2015年2期)2016-01-04 14:58:05
中药与临床(2015年5期)2015-12-17 02:39:28
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
食品工业科技(2014年5期)2014-03-11 18:14:08
