
Holistic analysis of Liuwei Dihuang pills using ultrasoniccell grinder extraction and ultra-performanceliquid chromatography
2017-01-09 11:56ZHAOTangjuanCHENJuanSHIYanping730000100049
色谱 2017年1期
ZHAO Tangjuan, CHEN Juan, SHI Yanping(1.,,,730000,; 2.,100049,)
Special issue for commemorating Professor ZOU Hanfa (Ⅱ)·Article
Holistic analysis of Liuwei Dihuang pills using ultrasoniccell grinder extraction and ultra-performanceliquid chromatography
ZHAO Tangjuan1,2, CHEN Juan1*, SHI Yanping1*
(1.KeyLaboratoryofChemistryofNorthwesternPlantResourcesofChineseAcademyofSciencesandKeyLaboratoryforNaturalMedicineofGansuProvince,LanzhouInstituteofChemicalPhysics,ChineseAcademyofSciences,Lanzhou730000,China; 2.UniversityofChineseAcademyofSciences,Beijing100049,China)
An efficient holistic analysis strategy was developed and validated for quality control of the traditional Chinese medicine (TCM) preparation, Liuwei Dihuang pills (LWDHPs). In this strategy, eight bioactive components, i. e. allantoin, morroniside, loganin, peoniflorin, acteoside, paeonol, alisol B 23-acetate and pachymic acid from the six medicinal herbs composing LWDHPs were selected as the evaluation markers, and then they were simultaneously extracted by an ultrasonic cell grinder extraction (UCGE) method and separated in a single run by an ultra-performance liquid chromatographic method coupled with photodiode array detection (UPLC-PDA). Response surface methodology (RSM), a multivariate experimental design technique, was used to optimize the UCGE parameters based on Box-Behnken design (BBD) model. The optimum extraction conditions were obtained at a ratio of solvent to solid 45 mL/g, extraction time of 40 min and extraction temperature of 20 ℃. Under such conditions, the higher yields of the eight analytes were acquired compared with the traditional extraction methods, indicating that UCGE was a fast, easy and efficient method for extracting bioactive constituents from LWDHPs. Chromatographic separation was achieved on an HSS T3 column (50 mm×2.1 mm, 1.8 μm) using a linear gradient elution of acetonitrile and water. Because of the different UV characteristics of these components, five detection wavelengths were used for quantitative analysis. The proposed method was validated through linearity, limits of detection, limits of quantification, precision, stability, repeatability, and accuracy. The validated method was applied to analyze LWDHPs, which provided a reference for the quality evaluation of LWDHPs.
ultrasonic cell grinder extraction (UCGE); response surface methodology (RSM); Liuwei Dihuang pills (LWDHPs); quality control
Traditional Chinese medicine (TCM), most of which are used as preparations, is playing an important role in health care, diseases prevention and treatment for centuries in China on account of their effective therapeutic performance, long historical clinical practice, affordability and few adverse effects[1,2]. With the excellent therapeutic effects, TCM has attracted worldwide attention and obtained the widespread applications [3]. Usually, TCM preparations are fairly complicated mixtures, which are composed of several herbal medicines, and each herbal medicine contains hundreds or even thousands of chemical constituents. Furthermore, the contents of chemical constituents presented in TCM preparations may vary significantly with the variations in raw materials from different geographic origins, climatic conditions, harvesting seasons and storages, pretreatments, and manufacturing processes. Thus, selecting favorable strategies for the quality consistency evaluation of TCM preparations is highly desired and extremely challenging.
Currently, one or more chemical markers from a certain composed herbal medicines are chosen to evaluate the quality of TCM preparations. However, it cannot accurately and comprehensively reflect the quality of TCM products, since therapeutic effect of a TCM preparation relies on the synergic effect of various chemical compositions from every herbal medicine composing a TCM preparation [1]. In recent years, chromatographic fingerprint technology based on the integrative and holistic properties of TCM has been developed and accepted [4,5]. It can offer the profiling of a complex system, but time-consuming process, special software and analytical methods are needed and thus have limited its extensive application [6]. In order to control the quality of TCM preparations more effectively, a new strategy, named as holistic analysis of multiple constituents from each component plant in a single run has been proposed by our group. Its core idea is that certain bioactive constituents from every constituent medicinal herb composing a TCM preparation are used as evaluation markers, and then they are simultaneously extracted, separated and quantified in a single run [7-10].
Liuwei Dihuang pills (LWDHPs), the well-known classical TCM prescription for “nourishing the kidney-yin”, are composed of Radix Rehmanniae Preparata, Rhizoma Dioscoreae, Fructus Corni, Cortex Moutan, Rhizoma Alismatis and Poria, and has been widely used for treating menoxenia, lowering blood sugar level, protecting the liver, reinforcing the function of the heart, curing disorder of immune and endocrine systems and inducing diuresis[11]. According to the Chinese Pharmacopoeia (2015 edition), three chemical markers, i. e. morroniside and loganin from Fructus Corni and paeonal from Cortex Moutan, are used as the assessment markers. But the quantitative information for the other four medicinal plants is lacking, thus such a strategy could not reflect the quality of LWDHPs adequately. In addition, the quantitative information of the above markers is obtained separately through two independent analytical procedures, which are complex and time-consuming. According to the results of pharmacology studies, pachymic acid from Poria [12,13], paeonol and peoniflorin from Cortex Moutan [14-17], acteoside from Radix Rehmanniae Preparata [18,19], allantoin from Rhizoma Dioscoreae [20,21], alisol B 23-acetate from Rhizoma Alismatis [22-25] and morroniside and loganin from Fructus Corni [26,27] are the main bioactive components from every component medicinal herb, respectively. Except for pachymic acid, peoniflorin and allantoin, these bioactive components are also the evaluation markers for the corresponding medicinal herbs in Chinese Pharmacopoeia (2015 edition). In order to obtain more comprehensive information for the quality control of LWDHPs, the eight bioactive components described above will be chosen as the evaluation markers in the subsequent study.
To obtain the bioactive components from LWDHPs, an efficient extraction methodology is indispensable. Conventional solvent extraction methods, such as Soxhlet extraction, reflux extraction and maceration extraction have been widely applied in TCM. In recent years, a novel extraction method, named as ultrasonic cell grinder extraction (UCGE) method has been proposed. Some outstanding advantages are involved in this method such as easy-to-use operation mode, fast extraction rate, high extraction efficiency and inexpensive apparatus [28-30]. During the UCGE process, the ultrasonic energy can induce the cavitation of the extraction solvent. Subsequently, the cavitation force of ultrasound successively accelerates the heat and mass transfer rate to disrupt plant cell walls and facilitates the release of required extractable compounds thus providing a desirable extraction yield [30]. In this work, the UCGE method was employed for the simultaneous extraction of the above eight components, and a comparative study on the extraction efficiency between UCGE and conventional solvent extraction methods was performed. In addition, response surface methodology (RSM), a multivariate experimental design technique, was used to optimize the UCGE parameters affecting the extraction efficiency based on Box-Behnken design (BBD) model.
To obtain good separation for all the markers in a single run, an appropriate analytical method is necessary. And according to our previous study, an ultra-performance liquid chromatographic (UPLC) method could fully meet this requirement owing to its superior performance over conventional HPLC method, such as increased peak capacity, improved resolution and shorter retention time. In this work, an UPLC method coupled with photodiode array detection (PAD) was developed for the simultaneous separation and quantification of the eight markers (pachymic acid, paeonol, peoniflorin, acteoside, allantoin, alisol B 23-acetate, morroniside and loganin) contained in LWDHPs. The chromatographic conditions and method validation are targeted in this procedure. In the end, the proposed extraction protocol and analytical method were applied for the quality control of six batches of LWDHPs from three manufacturers.
1 Experimental
1.1 Chemicals and materials
Allantoin (analyte 1, batch No. PS150918-02), morroniside (analyte 2, batch No. PS13032804), loganin (analyte 3, batch No. PS13031401), acteoside (analyte 5, batch No. PS12121103), alisol B 23-acetate (analyte 7, batch No. PS0387-0010) and pachymic acid (analyte 8, batch No. PS13101404) were purchased from Chengdu Push Bio-Technology Co., Ltd. (Chengdu, China). Peoniflorin (analyte 4, batch No. MUST-14081408) was purchased from Chengdu Must Bio-Technology Co., Ltd. (Chengdu, China). Paeonol (analyte 6, batch No. 0708-9704) was purchased from National Institutes for Food and Drug Control (Beijing, China). The purities of the above standards were all above 98% and their molecular structures are shown in Fig. 1. Six batches of LWDHPs were obtained from three manufacturers in China. Chromatographic-grade methanol and acetonitrile were purchased from Merck Co. (Darmstadt, Germany). Other chemicals of analytical grade were purchased from Tianjin Chemical Reagent Co. (Tianjin, China). Ultra-pure water was purified using an OKPVRE water ultrapure system (Shanghai, China).
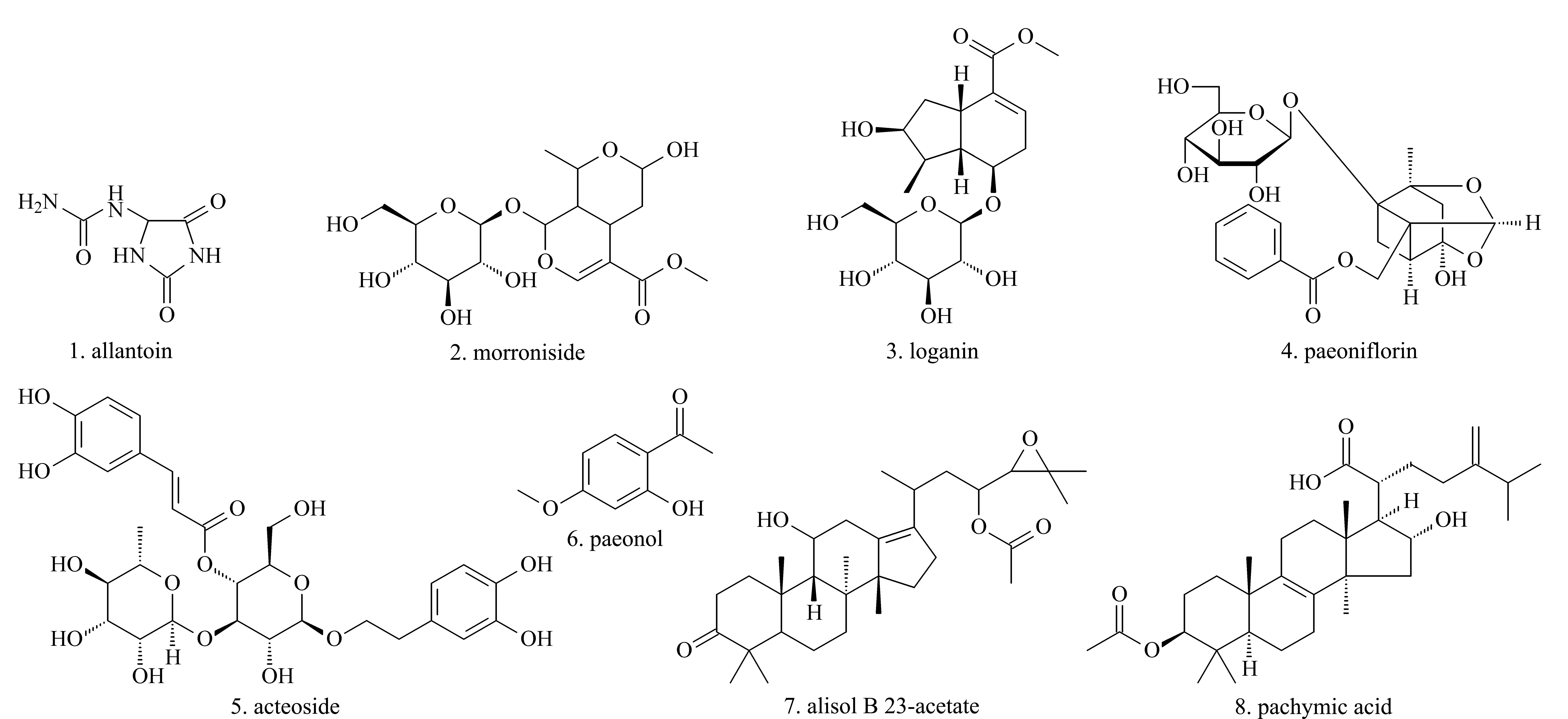
Fig. 1 Chemical structures of the analytes 1-8
1.2 Chromatographic system
UPLC analysis was performed on a Waters ACQUITY UPLCTMsystem (Waters, Milford, USA) equipped with a binary solvent delivery pump, an autosampler, a thermostated column compartment, a photodiode array detector and the Masslynx 4.1 workstation. Chromatographic separation was conducted on a Waters Acquity UPLC HSST3 column (50 mm×2.1 mm, 1.8 μm), using a linear gradient elution program of (A) acetonitrile and (B) water at a flow rate of 0.3 mL/min. The gradient elution program was as follows: 0-0.5 min, 0.1%A; 0.5-2.5 min, 0.1%A-18.0%A; 2.5-5.5 min, 18.0%A-68.0%A; 5.5-10.0 min, 68.0%A; 10.0-11.0 min, 68.0%A-100%A; 11.0-12.0 min, 100%A; 12.0-13.0 min, 100%A-0.1%A. The system was re-equilibrated with 0.1%A for 2 min before the next sample run. The temperatures of column and sample manager room were maintained at 30 ℃ and 18 ℃, respectively. The injection volume was 2 μL, and the partial loop with needle overfill was applied for sample injection. The absorption spectra of the compounds were recorded in the range of 190-400 nm (3D-plots were recorded), and the wavelengths used to measure the analytes were 210 nm for1, 7 and 8, 230 nm for 4, 240 nm for 2 and 3, 274 nm for 6, and 334 nm for 5.
1.3 Ultrasonic cell grinder system
Sample preparation was performed on a laboratory ultrasonic cell grinder (JY99-IID, Ningbo Scientz Biotechnology Co., Ltd., Ningbo, China), equipped with a generator, a converter, a booster horn and an alloy probe. The generator converts the main voltage to a high frequency of 21 kHz electrical energy, and then the converter transforms the electrical energy into 21 kHz mechanical energy. The booster horn, having the properties of thermally stable and corrosion resistant, could magnify the amplitude of ultrasonic. During the UCGE process, the ultrasonic energy focused on the alloy probe could give rise to sample powders suspending in solvent droplet adequately. Furthermore, abundant cavitating bubbles could generate heat flow and high-pressure shock wave, which increase the extraction efficiency.
1.4 Procedure
1.4.1Preparation of the standard solutions
The stock standard solutions of allantoin, morroniside, loganin, peoniflorin, acteoside, paeonol, alisol B 23-acetate and pachymic acid were prepared in methanol at the mass concentrations of 0.836, 0.944, 0.625, 1.024, 0.68, 1.37, 0.688 and 0.49 mg/mL, respectively. A mixed standard solution was prepared by adding 1.0 mL of each standard stock solution to a 10 mL volumetric flask and then diluting the contents to 10 mL with methanol. The calibration standard working solutions were freshly prepared by serially diluting the stock standard solutions to obtain the final concentrations. All the standard solutions prepared above were filtered through a 0.2 μm membrane before injection into the UPLC system and stored at 0-4 ℃ prior to analysis.
1.4.2Ultrasonic cell grinder procedure
After being crushed into small pieces, 1.0 g of LWDHPs was accurately weighed and introduced into a 150 mL beaker, and then extracted with 45 mL methanol in the ultrasonic cell grinder. The conditions for UCGE were as follows: cavitation time of 1.0 s, buffer time of 2.5 s, temperature of 20 ℃, power of 1 000 W, and the extraction time of 40 min. After cooling down to room temperature and making up for the lost weight, the extract was filtered through a qualitative filter paper and a 0.2 μm membrane filter successively and made ready for analysis. All the solutions were stored in a refrigerator at 0-4 ℃ prior to analysis. Furthermore, the comparisons of extraction efficiencies of UCGE with the other solvent extraction methods, including soxhlet extraction, reflux extraction, maceration extraction and ultrasonic extraction were made. The extraction yield (EY, mg/g) of the experimental evaluation markers was calculated based on the following equation:
EY=mi/mo
(1)
wheremi(mg) andmo(g) are the amount of the selected markers and the weight of LWDHPs prescription, respectively.
1.4.3Single factor experiment for UCGE
The effects of the independent factors involved in UCGE, including methanol concentration, ratio of solvent to solid, extraction time, extraction temperature, cavitation time, buffer time, and power on extraction yields of the selected markers were investigated by using a mono-factorial experimental design. The test ranges for different factor in preliminary experiment were carried out with the following presentations: methanol volume percentage (0%, 20%, 40%, 60%, 70%, 80%, 90%, and 100%), ratio of solvent to solid (5, 10, 20, 30, 40, 50, and 60 mL/g), extraction time (5, 10, 20, 30, 40, 50, and 60 min), extraction temperature (0, 10, 20, 30, 40, 50, and 60 ℃), cavitation time (0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 s), buffer time (0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 s), and power (800, 1 000, 1 200, 1 400, 1 600, and 1 800 W).
1.4.4Experimental design for the optimization of UCGE
RSM is a multivariate experimental design technique that is useful for modeling and analyzing engineering problems. BBD is one of the most common types of RSM models for operation optimization in drugs and food study. In this work, BBD was selected to optimize the extraction conditions through RSM since it requires fewer runs.
On the basis of the single-factor experimental results, a symmetrical three-level, three factorial, and three replicates of the central run, was applied to statistically optimize the selected markers extraction from LWDHPs prescription. The interaction and quadratic effects of three independent factors (X1, ratio of solvent to solid;X2, extraction time;X3, extraction temperature) on the yields of the extracts were evaluated. The experimental ranges for the selected factors were also confirmed based on the single-factor experimental results. In order to evaluate the importance of these factors conveniently, their levels were normalized and coded in comparable values. As shown in Table 1, each independent factor was coded at three levels between +1, 0, and -1 for high, intermediate and low level. For statistical calculation, the three variables were coded according to the following equation:
xi=Xi-Xo/ΔX
(2)
wherexi(i=1, 2, 3) is the coded value of the variable;Xiis the actual value of the variable;Xois the actual value of the variable at the center point;ΔXis the step change value of the variable.
Design Expert software (Trial version 8.0.6, Stat-Ease Inc., USA) was employed to analyze the experimental data, build and evaluate models, and predict the optimized conditions. A second-order polynomial model corresponding to the BBD was fitted to correlate the response (extraction yield, mg/g) with the independent variables according to the following regression model:
(3)
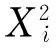

Table 1 Independent factors and three levels for BBD
1.4.5Evaluation of the analytical method
Method validation was performed in terms of linearity, limit of detection (LOD), limit of quantification (LOQ), precision, stability, repeatability, and accuracy according to the guidance for method validation for traditional Chinese medicines in Chinese Pharmacopoeia (Appendix X VIII A, 2010 edition).
2 Results and discussion
2.1 Optimization of UPLC conditions
In order to obtain a good resolution within a reasonable analysis time, the main chromatographic parameters were optimized. The UV spectra of the analytes between 190 and 400 nm were acquired using a photodiode array detector. Considering the sensitivity and simplicity of sample analysis, 210 nm was selected for analyzing allantoin, alisol B 23-acetate and pachymic acid, with 230 nm for peoniflorin, 240 nm for morroniside and loganin, 274 nm for paeonol, and 334 nm for acteoside.
Considering the chemical polarities of the analytes, it is needed to test a medium polarity column or a polarity column. In this study, a UPLC HSST3 column could result in better resolution and peak shape within a reasonable analysis time (within 10 min). The selection of the mobile phases was guided by the need to obtain good resolution of adjacent peaks within a relatively short analysis time. Different mobile phases (including methanol, acetonitrile and water) and modifiers (including acetic acid, formic acid and phosphoric acid) were investigated to improve the separation and the shapes of the peaks. According to the results of the preliminary studies, using methanol as the mobile phase or adding acetic acid and formic acid to mobile phase could not obtain good peak shape for alisol B 23-acetate and pachymic acid. In addition, when phosphoric acid was added into water as the modifier, allantoin could not be maintained on the chromatographic column and was eluted with the solvent peak. Acetonitrile and water without any modifiers showed the best results in terms of retention time and peak shape, and thus were selected as the mobile phases for UPLC analysis.
Increasing column temperature may result in a certain influence on the separation selectivity by decreasing the viscosity of the selected mobile phases. Higher temperature could bring shorter analysis time, but could not provide more efficient separation. In the work, the effect of column temperature in the range of 25-35 ℃ on the separation efficiency was investigated, and 30 ℃ was a suitable temperature for the measurement. In addition, different injection volumes (1, 2, 3, 4 and 5 μL) were examined and 2 μL was suitable. After many tests, excellent separation of the eight analytes was achieved in a single run and the typical chromatographic profiles obtained from a mixed standard solution and a real sample solution are shown in Fig. 2.
2.2 Comparison of extraction methods and single factor experiment for UCGE
The effect of extraction method, solvent and many other factors on the extraction efficiency of UCGE was investigated. The results are summarized in Fig. 3. Five different extraction methods including UCGE, reflux protocol, Soxhlet extraction, maceration and ultrasonication were employed separately to extract the target analytes from LWDHPs and their extraction yields were compared. As shown in Fig. 3a, the extraction efficiency of UCGE was equal to that of Soxhlet protocol, but was higher than those of the other methods. Finally, UCGE was selected as the preferred extraction method for its simplicity and novelty. Subsequently, single factor experiments were performed to determine the range of variables required to obtain a more realistic mode for the optimization of UCGE process by RSM. Extraction efficiency of different solvents including methanol, ethanol, acetonitrile, water, and aqueous methanol were evaluated and compared (Fig. 3b and 3c), and methanol was chosen as the extraction solvent. In addition, extraction efficiency of the ratio between solvent and solid, and the other main parameters were investigated. According to the results in Fig. 3d-3i, the extraction yields of the targets fluctuated barely with the variation of cavitation time, buffer time and power, while ratio of solvent to solid, extraction temperature and extraction time were confirmed as the main factors for the RSM with the appropriate ranges from 30 mL/g to 50 mL/g, 20 ℃ to 40 ℃ and 20 min to 40 min, respectively.

Fig. 2 UPLC chromatograms of (a) a mixed standard solution and (b) a real sample solutionPeaks: 1. allantoin; 2. morroniside; 3. loganin; 4. peoniflorin; 5. acteoside; 6. paeonol; 7. alisol B 23-acetate; 8. pachymic acid.
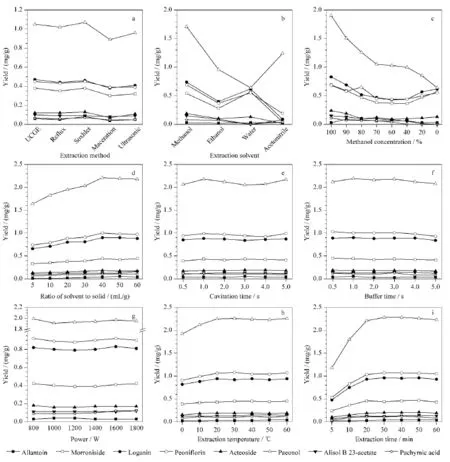
Fig. 3 Effects of (a) extraction method and (b-i) extraction parameters on the yields of analytes in single factor experiment
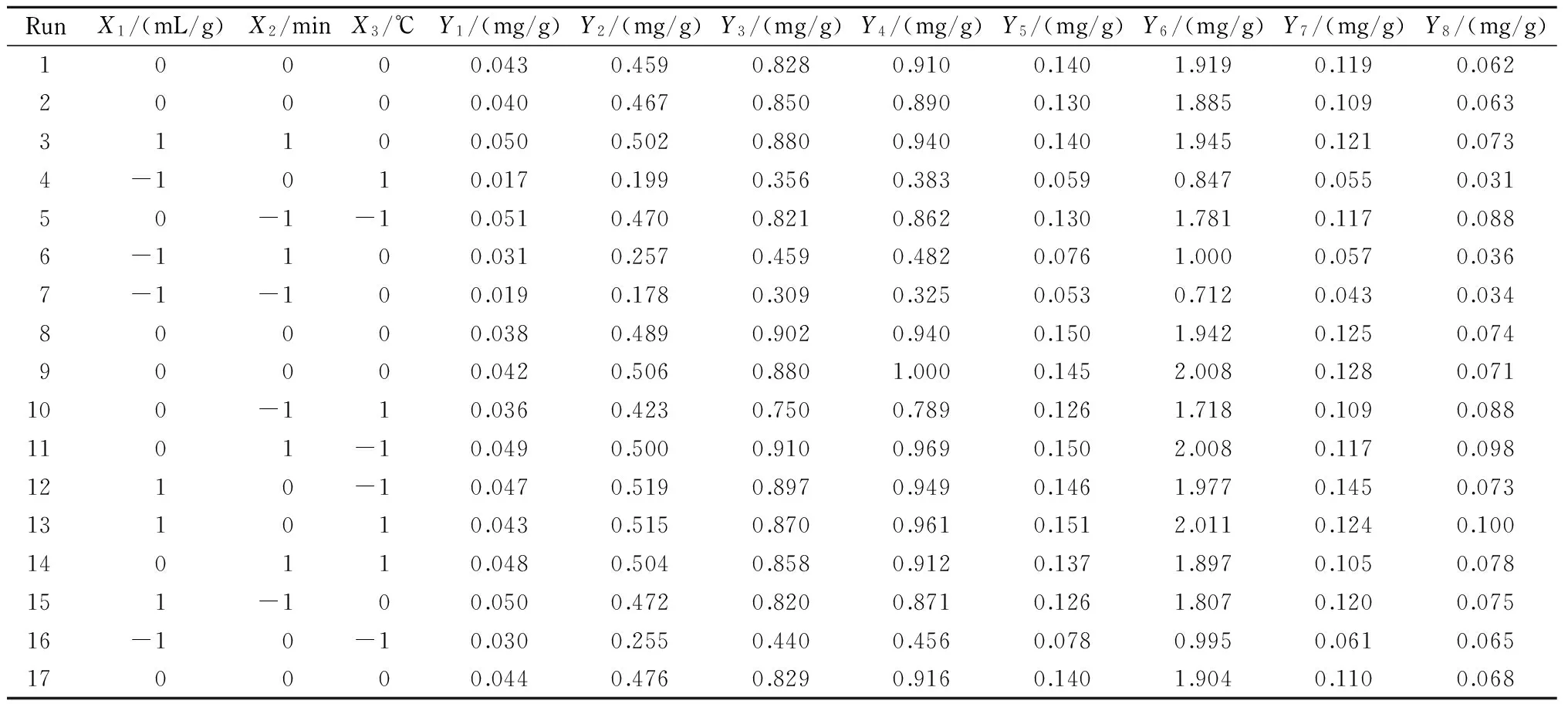
Table 2 Experimental runs of BBD and corresponding response values
2.3 Optimization of UCGE operating parameters by RSM
2.3.1Statistical analysis and model fitting of BBD
The complete experimental planning for BBD design consisted of 17 experimental points in random order with five replicates at the center point that are necessary for estimation of a pure error sum of squares. The design and experimental values are shown in Table 2. By employing multiple regression analysis on the experimental data, the predicted models addressed to the data, in terms of coded values, by the following second order polynomial equations:
Y1=0.041+0.012X1+0.002 75X2-0.004 125X3-0.003X1X2+0.002 25X1X3+0.003 5X2X3-0.007 825X21+0.003 925X22+0.000 675X23
(4)
Y2=0.48+0.14X1+0.028X2-0.013X3-0.012X1X2+0.013X1X3+0.013X2X3-0.11X21-0.012X22+0.007 3X23
(5)
Y3=0.86+0.24X1+0.051X2-0.029X3-0.023X1X2+0.014X1X3+0.004 75X2X3-0.22X21-0.023X22+0.000 35X23
(6)
Y4=0.93+0.26X1+0.057X2-0.024X3-0.022X1X2+0.021X1X3+0.004X2X3-0.24X21-0.04X22+0.007 725X23
(7)
Y5=0.14+0.037X1+0.008 5X2-0.003 875X3-0.002 25X1X2+0.006X1X3-0.002 25X2X3-0.035X21-0.007 5X22+0.002 25X23
(8)
Y6=1.93+0.52X1+0.1X2-0.036X3-0.038X1X2+0.046X1X3-0.012X2X3-0.48X21-0.086X22+0.005 45X23
(9)
Y7=0.12+0.037X1+0.001 375X2-0.005 875X3-0.003 25X1X2-0.003 75X1X3-0.001X2X3-0.024X21-0.008 6X22+0.002 4X23
(10)
(11)
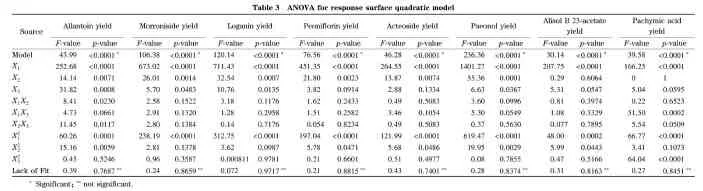
whereY1-Y8are the yields of allantoin, morroniside, loganin, peoniflorin, acteoside, paeonol, alisol B 23-acetate and pachymic acid, respectively;X1,X2andX3are the variables of ratio of solution to solid, extraction time and extraction temperature, respectively. The proposed models were evaluated by analysis of variance (ANOVA), and the results are shown in Table 3. Thep-value is used to estimate the significance of each coefficient, which suggests the interaction pattern between each independent variable. With the decrease ofp-values, the significance of the corresponding coefficient increases. In general, it is considered that the model terms were significant at ap-value of less than 0.05. The significance ofF-value depends on the number of degrees of freedom (DF) in the model and a very lowp-value with a high modelF-value (95% confidence level) indicated that the model is significant. The obtained modelF-values of 43.99, 106.38, 120.14, 76.56, 46.28, 236.36, 30.14, 39.58 imply the models were significant (p<0.000 1). There was only a 0.01% chance that a modelF-value this large could occur due to noise for each regression model. In these cases, the linear term of ratio of solvent to solid (X1) was the most significant model term.P-values greater than 0.100 0 indicate the model terms were not significant. The lack of fitF-values of 0.39, 0.24, 0.072, 0.21, 0.43, 0.28, 0.31, and 0.27 for allantoin, morroniside, loganin, peoniflorin, acteoside, paeonol, alisol B 23-acetate and pachymic acid imply the lack of fit was not significant relative to the pure error. There was a 76.87% chance that the lack of fitF-value this large could occur due to the noise for allantoin, while 86.59%, 97.17%, 88.15%, 74.01%, 83.74%, 81.63%, and 84.51% chances for morroniside, loganin, peoniflorin, acteoside, paeonol, alisol B 23-acetate and pachymic acid, respectively. It suggested that the yield of the eight compounds could be predicted by the model equations (4)-(11).
ItemR2AdjR2PredR2AdequateprecisionY10.98260.96030.916321.826Y20.99270.98340.972829.016Y30.99360.98530.985231.239Y40.98990.97700.964124.307Y50.98350.96220.915520.095Y60.99670.99250.986642.930Y70.97480.94250.891617.854Y80.98070.95590.923121.244
Furthermore, the fitness of the polynomial model equations could be confirmed by the determination coefficient (R2), and the values of adjusted determination coefficient (adjR2) of models were evaluated to check the model adequacies. The results of analytes of fit statistics for the response values are shown in Table 4. The values ofR2for the eight equations were all above 0.95, indicating that more than 95% of the variability in the response could be predicted by the models. The values of the adjR2(>0.90) also confirmed that the models were significant. The values of predicted determination coefficient (predR2) were in reasonable agreement with the values of adjR2. Adequate precision is used to measure signal to noise ratio, and a ratio greater than 4 is desirable. In this work, the values of ratio were found to be more than 17.0, which indicated adequate signals and that the models could be used to navigate the design space.
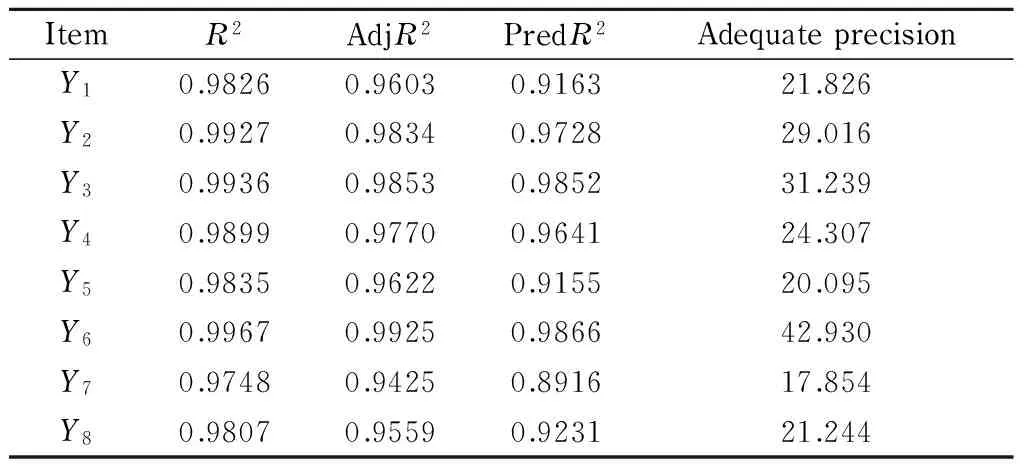
Table 4 Fit statistics for the response values

Fig. 4 Response surface 3D plots showing effects of ratio of solvent to solid (X1), extraction time (X2) and their reciprocal interaction on extraction yields of analytes
2.3.2Response surfaces analysis
The relationship between the responses and the experimental variables could be illustrated graphically to investigate the interactions of the variables and to determine the optimal level of each variable for the maximum response by plotting three-dimensional response surface plots. Each plot shows a pair of factors by keeping the other factor constant at its middle level.
Fig. 4 shows the effects of ratio of solvent to solid (X1), extraction time (X2) and their reciprocal interaction on extraction yields of the dependent variables (Y1,Y2,Y3,Y4,Y5,Y6,Y7andY8), while the extraction temperature (X3) is constant at 30 ℃. The results indicated thatX1was the major factor affecting extraction yield. WithX1increased, extraction yields of the eight dependent variables first increased rapidly and then changed slightly when the yields reached to peak, whereas the longerX2, the higher extraction yields of all the analytes. The higher extraction yields of the eight analytes could be obtained, whenX1was chosen in the range of 45-50 mL/g andX2was chosen in the range of 35-40 min.

Fig. 5 Response surface 3D plots showing effect of ratio of solvent to solid (X1), extraction temperature (X3) and their reciprocal interaction on extraction yields of analytes
Fig. 5 shows the effects ofX1,X3and their reciprocal effects on extraction yields of the eight dependent variables, whenX2is constant at 30 min. The extraction yields changed with the increase ofX1in a trend similar to Fig. 4. With increase inX3, the extraction yields of allantoin, loganin and alisol B 23-acetate (Fig. 5a, 5c and 5g) showed a slight decline, whereas a slight rise was found in the extraction yields of morroniside, peoniflorin, acteoside and paeonol (Fig. 5b, 5d, 5e and 5f). In addition,X3showed a trend to first decrease and then increase the extraction yield of pachymic acid (Fig. 5h).
Fig. 6 shows the effects ofX2,X3and their reciprocal effects on extraction yields of the eight dependent variables, whileX1is constant at 40. A higher extraction yield was acquired withX2in the range of 35-40 min andX3in the range of 20-25 ℃.X3showed a small effect on the extraction yields of allantoin, morroniside, loganin, peoniflorin, acteoside, paeonol and alisol B 23-acetate(Y1,Y2,Y3,Y4,Y5,Y6andY7), while it showed an inverted parabolic curve (first decrease and then increase) on the extraction yield of pachymic acid (Fig. 6h). With the increase ofX2, the extraction yields of allantoin, morroniside, loganin, peoniflorin (Fig. 6a, 6b, 6c and 6d) showed a slight escalating trend; however, for the extraction yields of acteoside, paeonol and alisol B 23-acetate (Fig. 6e, 6f and 6g), it showed a parabolic curve, and for the extraction yields of pachymic acid(Fig. 6h), it showed a gradually declined trend. WhenX3was at a low level,X2should be increased to get high extraction yields.
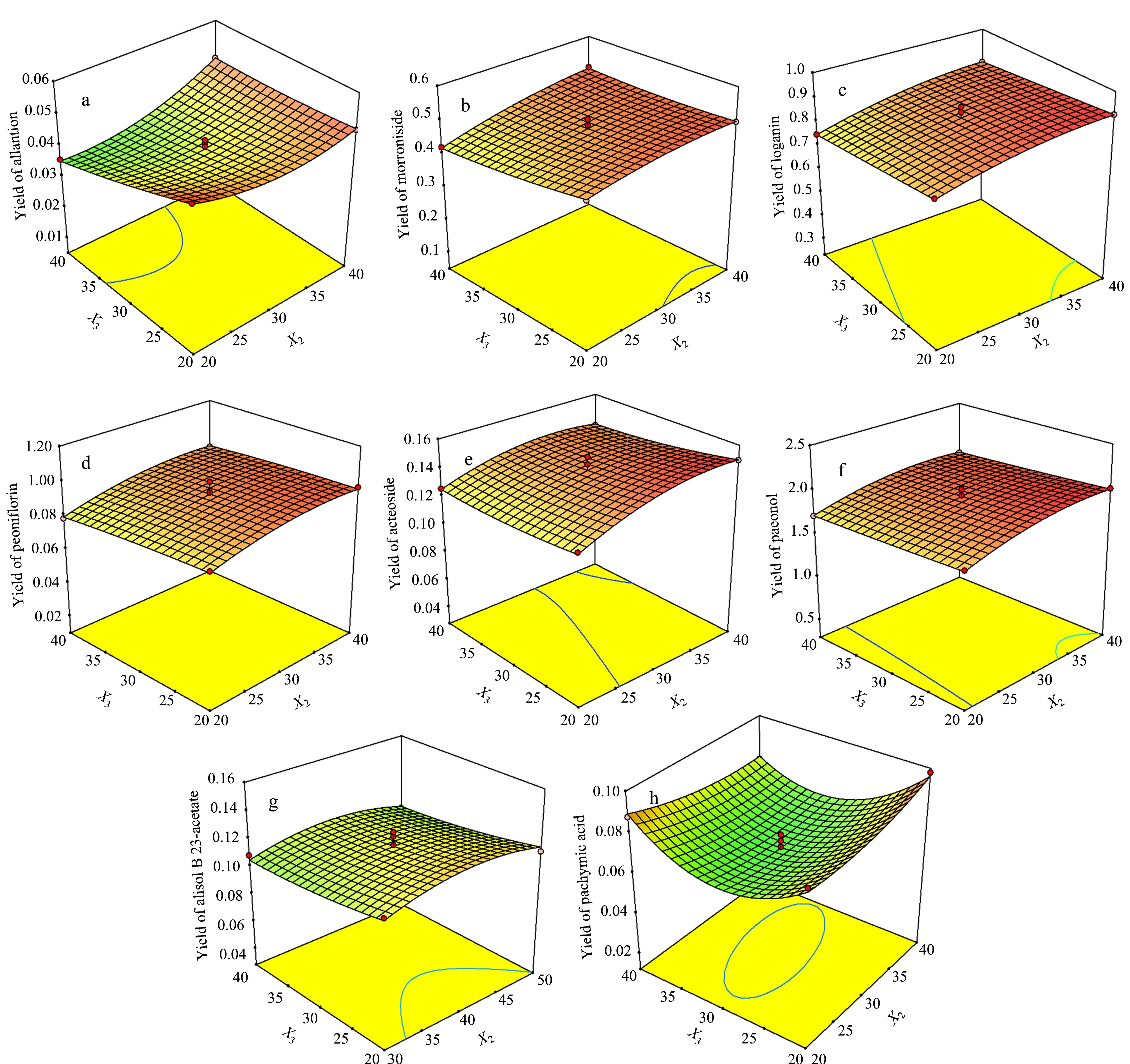
Fig. 6 Response surface 3D plots showing effects of extraction time (X2), extraction temperature (X3) and their reciprocal interaction on extraction yields of analytes
2.3.3Verification of optimized models
The verification experiments were conducted with the optimum extraction conditions (ratio of solvent to solid, 45 mL/g; extraction time, 40 min; and extraction temperature, 20 ℃. Three duplicate analyses were performed for experimental values. The experimental and predicted values of responses under optimum conditions are listed in Table 5. The relative standard deviation (RSD) values for the eight analytes ranged from 0 to 3.82%, indicating that the experimental values were in good agreement with the predicted ones. On the whole, the response models were adequate to reflect the optimization.
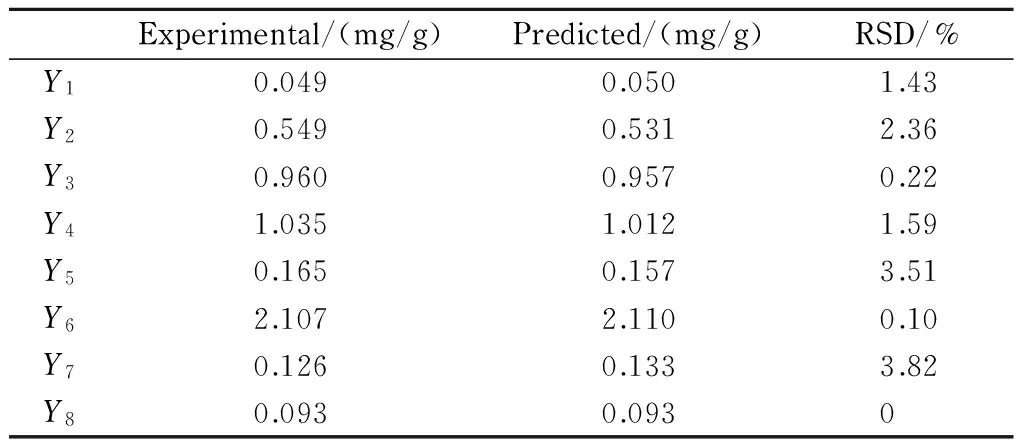
Table 5 Experimental and predicted values of responses under the optimum conditions (n=3)
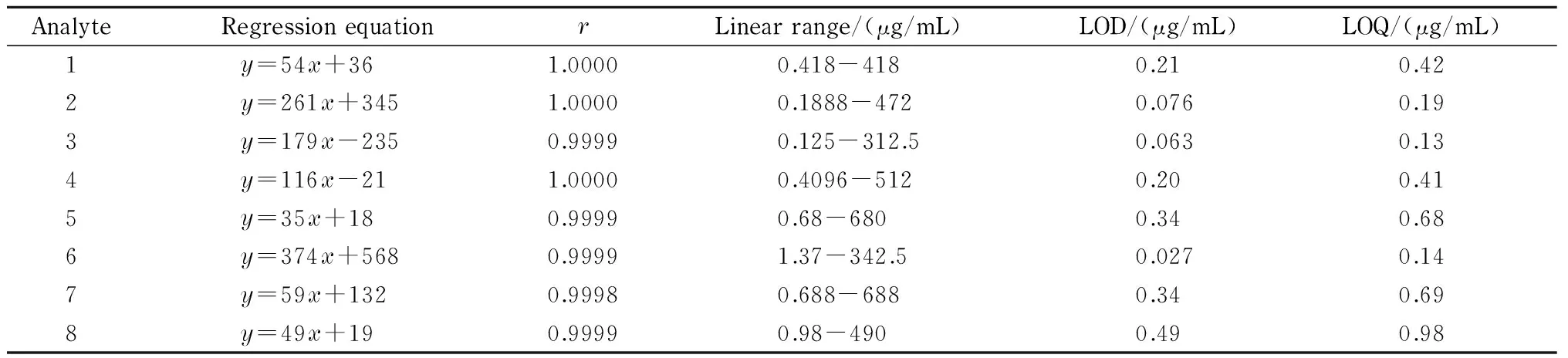
Table 6 Calibration plots, LODs and LOQs
y: peak area;x: mass concentration (μg/mL) of the analyte.
2.4 Method validation
2.4.1Calibration plots, LODs and LOQs
Under the optimal conditions, the calibration standard working solutions at six mass concentration levels were analyzed using UPLC method, and three duplicate analyses were performed for each level. The calibration curves were established by plotting the peak areas against the concentrations of analytes with linear regression analysis, and the regression equations are expressed asy=ax+b, whereyis peak area andxis mass concentration (μg/mL) of the analytes. Satisfactory correlation coefficients were obtained for all the analytes. LOD and LOQ were determined as the mass concentrations at the signal to noise ratios of 3 and 10, respectively. The regression equations, correlation coefficients (r), linear ranges, LODs, and LOQs are listed in Table 6.
2.4.2Precision, repeatability and stability
The precision of the developed method was evaluated by analyzing a mixed standard solution six times within a day for intra-day variation, and on six consecutive days for inter-day variation. The RSD values of the peak areas were 0.58%-1.17% for the intra-assay, while 0.98%-1.59% for the inter-assay precision, indicating the precision of the method was satisfactory.
Analysis repeatability was performed using the injections of six different sample solutions of LWDHPs (batch No. 14033623) which were prepared in parallel according to the procedure described above. The RSD values of the component content were 1.52%-4.33%, providing the accepted repeatability of the purposed method.
For the stability test, the same sample solutions were analyzed at 2 h intervals during storage for 12 h at room temperature. The sample solution can be regarded as stable within 12 h because the RSD values of the peak areas were 0.93%-2.65%.
2.4.3Accuracy
The accuracy of the method was investigated using a spiked recovery test with eight standards at three different mass concentration levels (low, medium, and high) in three replicates to assess matrix interferences. It is worth noting that the authentic standards of the analytes were separately added to an originally analyzed real sample (balch No. 14033623), for which the concentrations of the interested compounds were already known. Subsequently, three sets of spiked samples were treated according to the proposed procedure described above and analyzed by UPLC method. The recovery was used to evaluate the accuracy of the method, which was calculated by the following formula: recovery=found amount/added amount×100%. The recoveries of the analytes were calculated and summarized in Table 7. The average recoveries of the investigated components were in the range of 96.8%-100.6% with RSD values ranging from 0.7% to 3.8%, indicating that the proposed method enables highly accurate simultaneous analysis of the eight analytes in LWDHPs.

Table 7 Rusults of the recovery studies for determination of the eight analytes in LWDHPs (n=3)
2.5 Sample analysis
The developed method was applied to analyze the eight analytes in six batches of LWDHPs from three manufacturers. The contents of the eight components in the samples are presented in Table 8. It was shown that the contents of the analytes fluctuated substantially for different batches, as well as for different manufacturers. Paeonol, the main component in the six batches of LWDHPs, ranged from 0.90 to 2.31 mg/g. The large variation of specific component concentrations in the products manufactured by different factories is probably due to either different manufacturing processes or different sources of crude herbs, which demonstrates the importance and necessity of quality control once again. The developed analytical method appears to be an applicable tool for quantitative analysis of active components in LWDHPs and could serve as a reference for batch-to-batch quality control of LWDHPs.

Table 8 Contents of the eight analytes in LWDHPs from different production batches (n=3)
3 Conclusions
Efficient procedures to evaluate and control the quality of TCM preparations are urgently needed. The determination of multiple constituents from every component herb is the preferred strategy, which named holistic analysis strategy. In the work, the eight bioactive components from six medicinal herbs composing LWDHPs were selected as the evaluation markers. The extraction of them was conducted by a feasible and efficient UCGE method that could make the extraction yields higher and extraction time shorter compared with the traditional extraction methods. In addition, RSM as a useful tool was used to optimize the experimental variables (ratio of solvent to solid, extraction temperature and extraction time). The quantitative information of the analytes was obtained in a single run with the UPLC method. The developed and validated method was confirmed to be promising for improving the quality control of LWDHPs. The proposed strategy may provide a reference for the quality control of other TCM preparations with relatively simple prescriptions.
[1] Jiang Y, David B, Tu P F, et al. Anal Chim Acta, 2010, 657: 9
[2] Lai Y H, So P K, Lo S C L, et al. Anal Chim Acta, 2012, 753: 73
[3] World Health Organization. Bull World Health Organ, 2004, 82: 238
[4] Xie B G, Gong T, Tang M H, et al. J Pharmaceut Biomed, 2008, 48: 1261
[5] Razmovski-Naumovski V, Tongkao-on W, Kimble B, et al. Mode Tradit Chin Med Mater Med, 2010, 12: 99
[6] Tistaert C, Dejaegher B, Vander Heyden Y. Anal Chim Acta, 2011, 690: 148
[7] Chen J, Wang G Y, Shi Y P. Acta Chromatogr, 2009, 21: 341
[8] Chen J, Yang Y, Shi Y P. Biomed Chromatogr, 2011, 25: 1045
[9] Chen J, Sun J N, Song X Y, et al. Anal Methods, 2012, 4: 2989
[10] Zhou X J, Chen J, Li Y D, et al. J Chromatogr Sci, 2015, 53: 1786
[11] Bao J F, Xu H, Xu F. Lishizhen Medicine and Materia Medica Research, 2004, 15: 874
[12] Shah V K, Na S S, Chong M S, et al. J Biomed Res, 2015, 16: 84
[13] Zhao Y B, Xu B, Zan J F, et al. Chinese Journal of Information on Traditional Chinese Medicine, 2009, 16: 41
[14] Wu H, Zhu Z Y, Zhang G Q, et al. J Ethnopharmacol, 2009, 125: 444
[15] Yu K, Wang Y W, Cheng Y Y. J Pharmaceut Biomed, 2006, 40: 1257
[16] Fan X H, Ma T C, Shen X, et al. Chinese Traditional Patent Medicine, 2012, 34: 317
[17] Hu Y F, Xu G B. Anhui Medical and Pharmaceutical Journal, 2014, 18: 589
[18] Deng Z J, Liu R X, Li C Q, et al. Journal of Guangdong Pharmaceutical University, 2015, 31: 625
[19] Wei G D, Liu H, Wen X S, et al. Journal of Chinese Medicinal Materials, 2013, 36: 853
[20] Wen Q, Nie P, Ding Y, et al. Central South Pharmacy, 2014, 12: 169
[21] Zhang L, Liu Y H, Chen G N. J Chromatogr A, 2004, 1043: 317
[22] Lin W J, Xu R Q, Zhang Y M, et al. Traditional Chinese Drug Research & Clinical Pharmacology, 2015, 26: 30
[23] Meng Q, Chen X L, Wang C Y, et al. Toxicol Appl Pharm, 2015, 283: 178
[24] Hur J M, Chol J W, Park J C. Arch Pharm Res, 2007, 30: 1543
[25] Meng Q, Chen X L, Wang C Y, et al. Pharm Res, 2015, 32: 3688
[26] Zhao M, Tao J, Qian D, et al. J Chromatogr B, 2016, 1009/1010: 122
[27] He K, Song S H, Zou Z Y, et al. Phytother Res, 2016, 30: 283
[28] Wang G Y, Qi H Y, Shi Y P. J Sep Sci, 2010, 33: 1730
[29] Xie X Y, Chen F F, Yu J, et al. Int J Food Sci Technol, 2014, 49: 616
[30] Li Y, Fabiano-Tixier A S, Tomao V, et al. Ultrason Sonochem, 2013, 20: 12
超声细胞粉碎萃取结合超高效液相色谱用于六味地黄丸的整体分析
赵唐娟1,2, 陈 娟1*, 师彦平1*
(1. 中国科学院兰州化学物理研究所,中国科学院西北特色植物资源化学重点实验室/甘肃省天然药物重点实验室, 兰州 730000; 2. 中国科学院大学, 北京 100049)
针对中药复方制剂六味地黄丸的质量控制提出了一种多指标全药材整体分析新策略。从组成六味地黄丸的每一味药材中选取1~2个药效成分,即选取熟地黄中的毛蕊花糖苷、山茱萸中的莫诺苷和马钱苷、牡丹皮中的丹皮酚和芍药苷、山药中的尿囊素、茯苓中的茯苓酸、泽泻中的23-乙酰泽泻醇B作为六味地黄丸的质量控制指标成分,采用快速、简便、高效的超声细胞粉碎提取(UCGE)法对这8个指标成分进行同步提取。用响应曲面分析中的Box-Behnken模式对UCGE提取过程中对提取效率影响较大的料液比、提取时间和提取温度等参数进行了优化。得到最优的提取条件:料液比1∶45 (g/mL)、提取时间40 min、提取温度20 ℃。进一步利用超高效液相色谱-二极管阵列检测(UPLC-PAD)技术实现多指标成分在同一根色谱柱上的同步分离和检测,从而建立六味地黄丸的多指标全药材整体色谱。采用HSS T3色谱柱(50 mm×2.1 mm, 1.8 μm),以乙腈-水为流动相进行梯度洗脱,基于指标成分的紫外光谱特征选择5个检测波长同时检测,并进行了线性关系、检出限、定量限、精密度、稳定性、重复性、准确性等方法学验证。该研究为六味地黄丸质量标准的建立提供了依据,同时也为中药复方制剂提供了一种质量控制新模式和新理念。
超声细胞粉碎提取;响应曲面法;六味地黄丸;质量控制
10.3724/SP.J.1123.2016.08025
Foundation item: National Natural Science Foundation of China (Nos. 21375136, 21575150); Scholar Program of West Light Project, Chinese Academy of Sciences.
O658
: AArticle IC:1000-8713(2017)01-0032-15
*Received date: 2016-08-23
*Corresponding author. Tel: +86-931-4968121; Fax: +86-931-4968094; E-mail: chenjuan@licp.cas.cn (CHEN Juan), shiyp@licp.cas.cn (SHI Yanping).
猜你喜欢
中国科学院院刊(2023年2期)2023-02-27
中老年保健(2022年2期)2022-08-24
——李振声
干旱地区农业研究(2022年3期)2022-06-09
中老年保健(2021年8期)2021-08-24
基层中医药(2021年10期)2021-06-05
中山大学学报(自然科学版)(中英文)(2020年1期)2020-02-26
首都食品与医药(2017年7期)2017-10-20
中成药(2017年5期)2017-06-13
中国科学院院刊(2016年8期)2016-03-24
中国医药导报(2015年27期)2015-02-28

- 色谱的其它文章
- 基于高通量解析算法的复杂样品重叠气相色谱-质谱信号的快速分析
- 移液枪头式固相微萃取-高效液相色谱法测定细胞培养液中的4种生物碱
- 整体柱在线固相微萃取-高效液相色谱法分析爽肤水中痕量雌激素
- 化学衍生辅助液相色谱-串联质谱测定枇杷膏中的齐墩果酸和熊果酸
- Investigation of aromatic impurities in liquefied petroleumgas by solid-phase extraction sampling coupled withgas chromatography-mass spectrometry
- Quantification of intracellular adenosine 5′-triphosphateand its metabolites by high performance liquidchromatography analysis