
生物碱aaptamine的全合成研究
2017-01-04 12:24:36陈建新王瑾瑾李银辉卢爱党
河北工业大学学报 2016年2期
陈建新,王瑾瑾,李银辉,卢爱党
(河北工业大学 海洋科学与工程学院,天津 300130)
生物碱aaptamine的全合成研究
陈建新,王瑾瑾,李银辉,卢爱党
(河北工业大学 海洋科学与工程学院,天津 300130)
生物碱 aaptam ines具有广泛的生物活性,有着重要的医药研究价值.以高藜芦胺为起始原料,经酰化、Bischler-Napieralski反应、氧化、硝化得到6,7-二甲氧基-8-硝基异喹啉-1-甲醛 (9).化合物9经Henry反应,用叔丁醇钾作碱催化与硝基甲烷反应得到1-(6,7-二甲氧基-8-硝基异喹啉)-2-硝基乙醇(10),化合物10再在DMAP催化、乙酸酐作用下脱水成 (E)-6,7-二甲氧基-8-硝基-1-(2-硝基乙烯基)异喹啉 (11),最后铁粉还原关环成功合成了aaptam ine,总收率为28%.aaptamine盐酸盐结构经1HNMR、13CNMR和HRMS表征确认,其他中间体结构均经1H NMR、13CNMR表征确认.
生物碱;aaptam ine;全合成;Henry反应;催化脱水
0 引言
海洋是人类物质资源的天然宝库,已知海洋生物的物种总数占地球生物的80%以上[1],从海洋天然产物中寻找新型活性药物先导已成为当今研究的热点.Aaptam ine(1)及其天然同系物统称为aaptam ines,是具有苯并[de][1,6]萘啶骨架的海洋生物碱.自1981年,Nakamura等人首次从日本冲绳县海域的海洋海绵生物中提取出aaptam ine(1)[2],其它结构的aaptam ines如:isoaapatam ine(2)、9-demethyl-aaptam ine(3)[3]及其二聚体[4]等同系物也相继被报道.近几十年来,世界各国对aaptam ines及其衍生物的应用展开了广泛的研究,逐渐发现该类生物碱除了清除自由基和抗氧化性外,还具有更广泛的药理作用,如抗癌、抗菌消炎、抗HIV病毒、抗寄生虫、抗抑郁、防污功能等作用[3,5],此外还有蛋白酶抑制活性[6].
Joule等人[7-8]在Tollari报道的合成路线[9]基础上进一步改进,从原料6,7-二甲氧基-1-甲基异喹啉出发,经浓硝酸硝化(41%)、二氧化硒氧化(54%)后生成6,7-二甲氧基-8-硝基异喹啉-1-甲醛,再与硝基甲烷在三氧化铝的作用下发生Henry反应生成硝基醇化合物(84%),然后在三氯化铝作用下连续脱水-分水得到硝基烯烃化合物(36%),最后用铁粉处理得到生物碱aaptam ine盐酸盐(89%).该路线共5步,总收率约为6%.
在前期工作的基础上[10-11],积累了大量制备硝基烯烃的经验,针对Joule合成路线中存在的缺陷进行改进,以高藜芦胺为原料制备天然产物生物碱 aaptam ine.通过硝化反应的调整、关键中间体实验方法的改进及条件探索,反应方程式如图2所示,探索出一条操作简便、收率较高的合成路线.从高藜芦胺出发,共需8步反应,总收率为28%;从6,7-二甲氧基-1-甲基异喹啉出发,共需5步反应,总收率为34%.
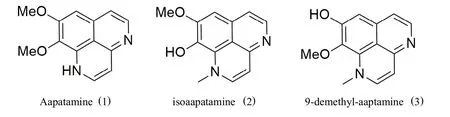
图1 Aaptamine及其同系物的化学结构Fig.1 Chemicalstructuresof aaptamine(1)and some congeners
1 实验部分
1.1 试剂与仪器
试剂:高藜芦胺(97%,南京多点化工),10%钯碳(南京多点化工),乙酸酐、三乙胺、DMAP、叔丁醇钾等均为分析纯(天津市河东区广达试剂).
仪器:Avance IIIplus 400MHz核磁共振仪,瑞士Bruker公司(TMS作内标);7.0 T傅立叶变换离子回旋共振质谱仪(美国IonSpec);X-4数字显微熔点仪,河南巩义予华仪器有限公司;CJB-S磁力搅拌器,河南巩义予华仪器有限公司.
1.2 实验方案
天然产物生物碱aaptam ine全合成路线如图2所示.
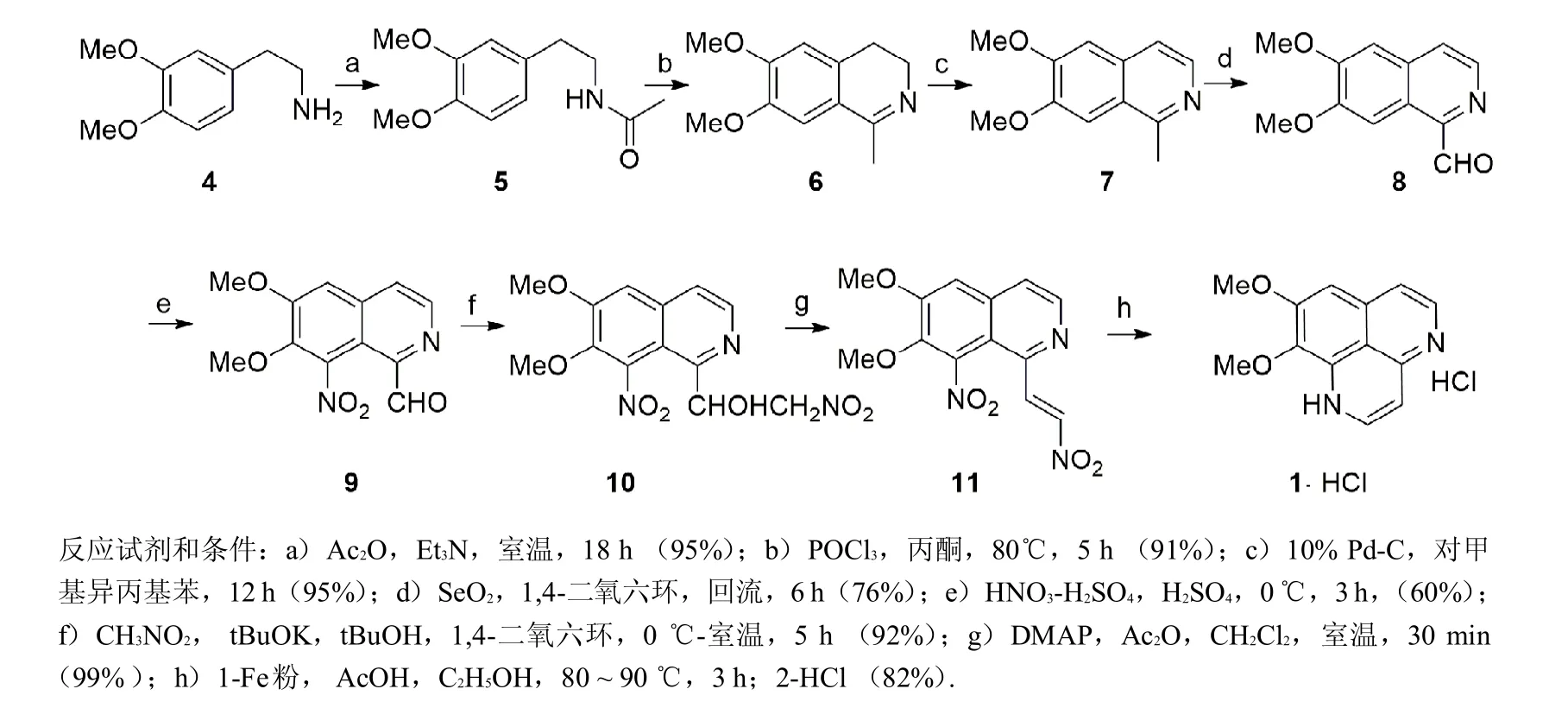
图2 Aaptamine的全合成路线Fig.2 TotalSynthesisof Aaptamine
1.3 实验方法
1.3.1 N-3,4-二甲氧基苯乙基乙酰胺 (5)的合成[12]
N2保护下,将乙酸酐(10.2 g,0.1mol)溶于二氯甲烷(200m L)中,然后滴加高藜芦胺 (4)(18.1 g,0.1mol).室温搅拌24 h后,依次加入1M HCl(100m L×2)、饱和NaHCO3(100m L)溶液和饱和氯化钠溶液(100m L)洗涤.有机相用无水硫酸镁干燥,过滤后真空脱溶得白色固体,乙醚重结晶提纯.收率95%;熔点:93~95℃(lit.[12]100~102℃);1HNMR(CDCl3,400MHz): 1.94(s,3H),2.75(t,J=7.2Hz,2H),3.48(dd,J=6.8Hz and 12.8Hz,2H),3.85(s,3H),3.86(s,3H),5.59(brs,1H),6.71(s,1H),6.73(d,J=2.0Hz,1H),6.80(d,J=8.0Hz,1H);13CNMR(CDCl3,100MHz):170.0,149.0,147.6,131.3,120.6,111.8,111.3,55.8,40.7,35.1,23.3.
1.3.2 6,7-二甲氧基-1-甲基-3,4-二氢异喹啉 (6)的合成[12]
N2保护下,将化合物5(11.6 g,50mmol)与POCl3(6.9m L,75mmol)溶解在100m L甲苯中,加热回流6h.冷却室温后过滤,然后将滤饼加入到盛有二氯甲烷(100m L)的圆底烧瓶中,用饱和K2CO3(100mL)调节pH=9~10,同时溶液由浑浊变澄清.混合液分液后,水相用二氯甲烷(50m L×3)萃取,合并有机相,用饱和食盐水洗涤,然后用无水硫酸钠干燥,过滤后真空脱溶,得棕色固体,柱层析(硅胶:100~200m,洗脱液为乙酸乙酯)提纯得浅棕色固体产物.收率:91%;熔点:97~99℃(lit.[12]124~126℃);1HNMR (CDCl3,400MHz): 2.36(s,3H),2.63(t,J=10.4Hz,2H),3.62(t,J=9.6Hz,2H),3.90(s,3 H),3.91(s,3 H),6.68(s,1 H),6.98(s,1H);13C NMR(CDCl3,100MHz):163.7,150.7,147.3,131.0,122.3,111.1,108.9,56.1,55.8,46.8,25.6,23.3.
1.3.3 6,7-二甲氧基-1-甲基异喹啉 (7)的合成[13]
将化合物6(12.2 g,59.3mmol)溶解在对异丙基甲苯(120m L)中,加入Pd/C(10%,1.0 g),反应体系在176℃温度下反应30 h.体系冷却室温后过滤除去钯碳,滤液真空脱溶后柱层析提纯(硅胶:100~200m,洗脱液为乙酸乙酯:甲醇=10:1),得棕色固体产物7.收率:95%;熔点:88~93℃(lit.[14]104 ~105℃);1HNMR(CDCl3,400MHz): 2.85(s,3H),3.98(s,3H),4.00(s,3H),7.00(s,1H),7.21(s,1H),7.34(d,J=7.6Hz,1H),8.24(d,J=7.6Hz,1H);13CNMR(CDCl3,100MHz):155.4,152.1,149.4,140.3,132.2,122.7,117.9,104.8,103.3,55.6,55.5,22.0.
1.3.4 6,7-二甲氧基异喹啉-1-甲醛 (8)的合成[8]
N2保护下,将SeO2(1.66 g,15mmol)溶解在1,4-二氧六环中(30m L),加热至95℃时滴加化合物7 (2.0 g,10 mmol)的1,4-二氧六环(60 m L)溶液.滴加完毕后,反应体系继续反应6 h.将体系冷却至室温,然后过滤,滤液真空脱溶.残留物柱层析提纯(硅胶:100~200m,洗脱液为石油醚:二氯甲烷:乙酸乙酯=1∶1∶1),提纯后提到淡粉色固体产物8.收率为76%;熔点:169~171℃(lit.[8]174~176℃);1HNMR(CDCl3,400MHz): 4.06(s,3H),4.10(s,3H),7.14(s,1H),7.74(d,J=5.6Hz,1H),8.63(d,J=5.2Hz,1H),8.75(s,1H),10.37(s,1H);13CNMR(CDCl3,100MHz):196.5,153.2,152.9,147.3,141.6,134.4,124.0,123.2,104.6,103.5,56.3,56.1.
1.3.5 6,7-二甲氧基-8-硝基异喹啉-1-甲醛 (9)的合成
0℃条件下,将化合物8(0.63g,2.89mmol)溶解在浓硫酸(5m L)中,缓慢滴加2M硝酸-硫酸溶液(1.45m L,1.0 equiv.).反应3 h后,缓慢滴加至冰水中,混合液用,NaOH(25%)溶液调节至pH=9.用二氯甲烷(60m L×3)萃取水相,合并有机相并用无水MgSO4干燥,过滤后真空脱溶得粗品,柱层析(硅胶:100~200 m,洗脱液石油醚∶二氯甲烷∶乙酸乙酯=1∶2∶2)提纯得到黄色固体9.收率为60%;熔点:194~196℃(lit.[8]203~205℃);1HNMR(CDCl3,400MHz): 4.09(s,3H),4.10(s,3H),7.30 (s,1H),7.75(d,J=5.6 Hz,1 H),8.65(d,J=5.2 Hz,1 H),10.12(s,1 H);13C NMR(CDCl3,100MHz):191.3,155.8,150.0,145.7,142.6,140.7,135.0,123.0,112.9,108.5,63.0,56.6.
1.3.6 1-(6,7-二甲氧基-8-硝基异喹啉)-2-硝基乙醇 (10)的合成
将化合物9(1.31g,5mmol)、CH3NO2(0.61 g,10mmol)溶解在tBuOH(5m L)和1,4-二氧六环(5mL)的混合溶剂中,0℃温度下,加入tBuOK(0.03mg,0.06equiv.).TLC检测,原料消失后加入H2O(25m L)和二氯甲烷(50 m L).混合体系分液后,水相用二氯甲烷(50 m L)萃取.合并有机相,用无水MgSO4干燥,过滤后真空脱溶,得到淡粉色固体硝基醇化合物10,用乙醚洗涤后得纯品.收率为92%;熔点:154~156℃(lit.[8]158~160℃);1H NMR(CDCl3,400MHz): 4.04(s,3H),4.10(s,3H),4.51(dd,J =8.8 and 11.2Hz,1 H),4.73(d,J=12.8Hz,1H),5.05(br s.1H),5.64(d,J=6.4Hz,1H),7.30 (s,1H),7.64(d,J=4.0Hz,1H),8.49(d,J=4.8Hz,1H);13CNMR(DMSO-d6,100MHz):154.7,153.6,143.9,141.8,141.0,135.6,121.2,113.0,110.8,79.8,69.1,63.1,57.4.
1.3.7 (E)-6,7-二甲氧基-8-硝基-1-(2-硝基乙烯基)异喹啉 (11)的合成
4.06(s,3 H),4.10(s,3 H),7.67(d,J=5.6 Hz,1 H),7.93(d,J=12.8 Hz,1 H),8.13(d,J= 12.8Hz,1H),8.56(d,J=5.2Hz,1H);13CNMR(DMSO-d6,100MHz):159.0,148.1,147.8,147.8,147.6,143.7,139.2,137.5,126.8,118.2,114.5,67.2,61.6.
1.3.8 Aaptamine盐酸盐的合成
N2保护下,将化合物11(30 mg,0.1 mmol)溶解在乙酸(2.5 m L)和乙醇(2.5 m L)的混合溶液中,一次性加入铁粉(140mg).反应液室温搅拌2 h后加热至80~90℃,继续反应0.75 h.反应体系冷却至室温,快速抽滤,向滤液中加入浓盐酸(0.5m L),室温搅拌30m in后真空脱溶,残留物柱层析快速提纯(酸性氧化铝:200~300m,洗脱液二氯甲烷∶甲醇=10∶1)得棕色固体Aaptam ine盐酸盐,再用三氯甲烷和丙酮重结晶得纯品.收率为82%;熔点:110~112℃(lit.[8]108~112℃);1HNMR(DMSO-d6,400MHz):3.82(s,3 H),3.99(s,3 H),6.51(d,J=6.8Hz,1H),6.85(d,J=6.8Hz,1H),7.13(s,1H),7.41(d,J=6.8Hz,1H),7.86(t,J=6.0Hz,1H),12.31(s,1H),13.24(s,1H);13CNMR(DMSO-d6,100MHz):157.4,150.2,142.4,134.2,133.1,131.9,130.2,116.8,113.1,101.4,98.5,60.9,57.0;HRMS(ESI)calcd for C13H13ClN2O2(M+Na-HCl)+251.079 1,found 251.079 2.
2 结果与讨论
2.1 化合物9的合成条件探索
按照文献 [8]报道,化合物7用二氧化硒氧化能顺利得到相应的醛8;但如果以原料1-甲基-6,7-二甲氧基-8-硝基异喹啉进行氧化,反应将进行的异常困难,不仅使用的溶剂(1,4-二氧六环)要绝对的无水,反应时间也大大延长,而且伴随有副产物6-甲氧基-7-羟基-8-硝基异喹啉-1-甲酸的生成.为了避免以上情况的发生,在合成过程中首先将化合物7进行氧化以76%的收率得到相应的醛8,然后将化合物8溶解在浓硫酸中,0℃条件下滴加2M HNO3的浓硫酸溶液,以60%的收率得到相应的硝基化合物.
2.2 关键中间体硝基醇化合物10的合成条件探索
在前期工作基础上,发现叔丁醇钾催化作用下,化合物9与硝基甲烷能顺利发生Henry反应生成相应的硝基醇化合物10.将碱及其用量、温度、溶剂等因素对反应转化率的影响进行对比实验,反应进行12 h后处理,结果见表1.
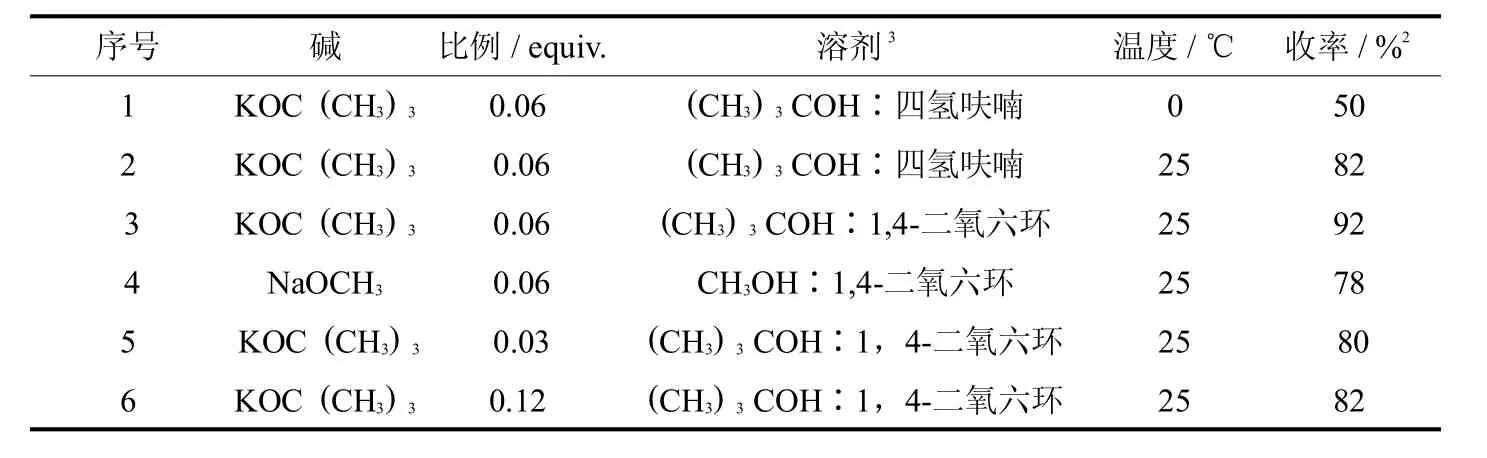
表1 不同因素对硝基醇10制备的影响Tab.1 The exploration of rection conditions for the synthesisof alcohol101
表1实验结果表明:为了降低硝基醇化合物的脱水现象,首先在0℃下进行实验,溶剂采用叔丁醇和四氢呋喃(体积比为1∶1),化合物10的收率只有50%(序号1),反应过程中检测存在有大量原料.将体系的温度升至室温25℃下进行,收率大幅度提高至82%(序号2),反应过程中仍然发现因溶解问题,部分原料未能参加反应.根据实验结果对反应中使用的溶剂进行调整,采用叔丁醇和1,4-二氧六环(体积比为1:1),反应物加入后搅拌半小时左右既能全部溶解,收率提高至92%(序号3).使用甲醇钠催化剂或者降低叔丁醇钾用量时,反应收率大幅下降,原料反应不充分(序号4和序号5);但若提高叔丁醇钾的用量,柱层析分离过程中得到15%的脱水产物.
2.3 硝基烯烃11的合成条件探索
根据文献报道,硝基醇脱水的过程比较困难,分子中如果有邻近的氮原子、氧原子很容易发生脱水-重排[8,15-16](见图3),导致副产物1-甲基-6,7-二甲氧基-8-硝基异喹啉生成.为了克服副产物的生成,采用碱催化作用下与乙酸酐首先发生酯化反应,进而脱除一分子乙酸得到相应的硝基烯烃11.
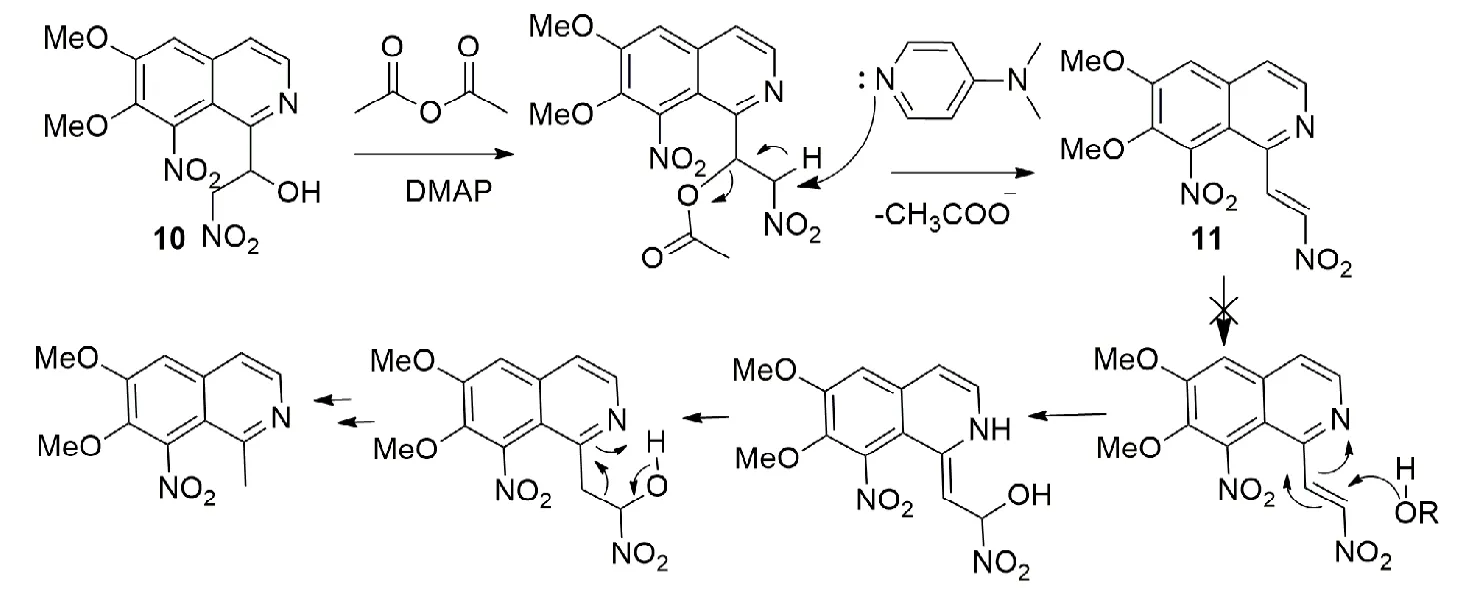
图3 化合物9催化脱水过程Fig.3 The catalytic dehydrogenation processof alcohol9
为了考察碱的种类及用量、反应温度等因素对化合物11收率的影响,以硝基醇化合物10、乙酸酐为原料,对以上因素进行筛选.
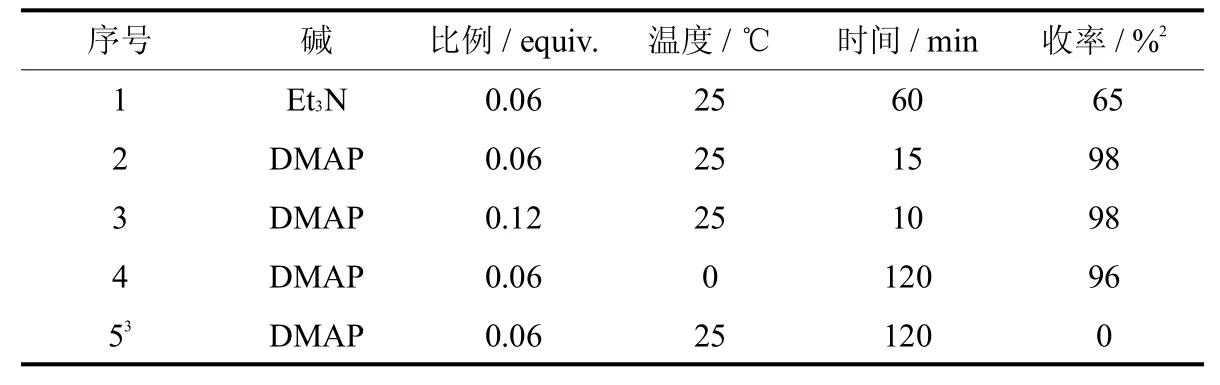
表2 化合物11合成条件的探索Tab.2 The exploration of rection conditions for the synthesisof nitroolefin 111
从表2结果可知,当使用碱为三乙胺(6%equiv.)时,化合物11的收率只有65%,有大量原料未完全反应;使用DMAP(6%equiv.)为催化剂时,室温条件下,反应15m in原料就能完全反应,几乎以定量的收率得到化合物11;增加DMAP用量为12%equiv.反应时间缩短;降低反应温度为0℃时,反应时间大大延长.无论是温度和DMAP用量改变,主要影响到反应的时间,对化合物11的收率并未有明显影响.但是如果反应中不添加乙酸酐(序号5),反应不能进行.
3 结论
以高藜芦胺为原料,对天然产物生物碱aaptam ine的全合成进行了优化,特别是对重要中间体10和11合成进行了优化.从碱的种类及其用量、反应温度、反应溶剂等方面优化后以92%的收率得到化合物10.DMAP催化作用下,硝基醇化合物与乙酸酐反应后以几乎定量收率得到硝基烯烃化合物11.经优化后,发展了一条合成生物碱aaptam ine的路线,从高藜芦胺出发,共需8步反应,总收率为28%.
[1]Staley A L,RinehartK L.Spectomycins,new antibacterialcompoundsproducedby Streptomycesspectabilis.Isolation,structures,and biosynthesis [J].JAntibiot(Tokyo),1994,47(12):1425-1433.
[2]Nakamura H,Kobayashi J,OhizumiY,etal.Isolation and structureof aaptaminea novelheteroaromatic ubstance possessing -blocking activity from the sea sponge Aaptosaaptos[J].Tetrahedron Lett,1982,23(52):5555-5558.
[3]LarghiEL,BohnM L,Kaufman TS.Aaptamineand related products.Theirisolation,chemicalsyntheses,and biologicalactivity[J].Tetrahedron,2009,65(22):4257-4282.
[4]Liu C L,Tang X L,LiPL,etal.Suberitine A-D,Fournew cytotoxic dimeric Aaptam inealkaloids from themarine sponge Aaptossuberitoides.Org Lett,2012,14(8):1994-1997.
[5]Dyshlovoy SA,Naetheth I,Venz S,etal.Proteomic profilingofgerm cellcancercells treatedw ithaaptam ine,amarinealkaloidw ithantiproliferative activity[J].JProtRes,2012,11(4):2316-2330.
[6]Tsukamoto S,Yamanokuchi R,Yoshitom i M,et al.Aaptamine,an alkaloid from the sponge Aaptos suberitoides,functions as a proteasome inhibitor[J].Bioorg Med Chem Lett,2010,20(11):3341-3343.
[7]BalczewskiP,MallonM K,Street JD,etal.A synthesisofaaptamine from 6,7-dimethoxy-1-methylisoquinoline[J].Tetrahedron Lett,1990,31(4):569-572.
[8]BalczewskiP,MallonM K,Street JD,etal.A synthesisofaaptm ine from 6,7-dimethoxy-1-methylisoquinoline[J].JChem Soc,Perkin Trans,1990,(11):3193-3198.
[9]BassoliA,MaddinelliG,Rindone B,etal.A simple synthesisof aaptam ine,a1H-benzo[de][1,6]-naphthyridinealkaloid[J].J Chem Soc Chem Commun,1987,(3):150-151.
[10]Lu AD,Hu K L,Wang Y M,etal.Enantioselectivesynthesisof trans-dihydrobenzofuransviaprimary am ine-thioureaorganocatalyzed intramolecular M ichaeladdition[J].JOrg Chem,2012,77(14):6208-6214.
[11]Lu AD,Liu T,Wu RH,etal.ARecyclableorganocatalystforasymmetricM ichaeladditionofacetone tonitroolefins[J].JOrg Chem,2011,76(10):3872-3879.
[12]SoniR,Cheung FK,Clarkson G C,etal.The importance of the N-H bond in Ru/TsDPEN complexes for asymmetric transfer hydrogenation of ketonesand im ines[J].Org BiomolChem,2011(9):3290-3294.
[13]FunabashiK,RatniH,KanaiM,etal.Enantioselective construction of quaternary stereocenter through a Reissert-type reaction catalyzed by an electronically tuned bifunctionalcatalyst:efficientsynthesisofvariousbiologically significantcompounds[J].JAm Chem Soc,2001,123(43):10784-10785.
[13]Dong J,ShiX X,Yan JJ,etal.Efficientand practicalone-potconversionsof N-tosyltetrahydroisoquinolines into isoquinolinesand of N-tosyltetrahydro--carbolines into-Carbolines through tandem -elimination and aromatization[J].Eur JOrg Chem,2010(36):6987-6992.
[14]Ahamed M,Thirukkumaran T,LeungW Y,etal.Aza-Henry reactionsof3,4-dihydroisoquinoline.Eur JOrg Chem,2010(31):5980-5988.
[15]Denmark SE,KeslerBS,Moo YC.Inter-and intramolecular[4+2]cycloadditionofnitroalkenesw itholefins2-nitrostyrenes[J].JOrg Chem,1992,57(18):4912-4924.
[责任编辑 田 丰 夏红梅]
Totalsynthesisof aaptam ine
CHEN Jianxin,WANG Jinjin,LIYinhui,LU Aidang
(SchoolofMarine Scienceand Engineering,HebeiUniversity of Technology,Tianjin 300130,China)
Aaptam inesalkaloidsw ith diversebiologicalpropertieshave importantpotential in drug development.6,7-Dimethoxy-8-nitroisoquinoline-1-carbaldehyde(9)wasobtained from homoveratrylam ineby acylation,Bischler-Napieralski reaction,oxidation and nitration.Compound 9wascondensedw ith nitromethaneusing tBuOK asbaseviaHenry reaction togive1-(6,7-dimethoxy-8-nitroisoquinolin-1-yl)-2-nitroethanol(10),and then dehydration in thepresentofAc2O using DMAPascatalyst.Iron-acetic acid treatmentof(E)-6,7-dimethoxy-8-nitro-1-(2-nitrovinyl)isoquinoline(11)produced aaptam ine.The totalyieldwas28%.Theaaptam inewasconfirmed by1HNMR,13CNMR and HRMS,and theother intermediateswere confirmed by1H NMR,13CNMR.
A lkaloids;aaptam ine;Totalsynthesis;Henry reaction;Catalytic dehydration
O621.3
A
1007-2373(2016)02-0044-06
10.14081/j.cnki.hgdxb.2016.02.008
2015-03-24
国家自然科学基金(21302038,21476059);河北省自然科学基金(B2013202237)
陈建新(1969-),男(汉族),教授,博士,博士生导师.通讯作者:卢爱党(1982-),女(汉族),讲师,博士.
数字出版日期:2016-04-26 数字出版网址:http://www.cnki.net/kcms/detail/13.1208.T.20160426.0939.004.htm l
猜你喜欢
中国野生植物资源(2024年2期)2024-03-19 05:55:38
合成纤维工业(2023年5期)2023-10-31 13:28:02
化工技术与开发(2022年4期)2022-04-22 09:10:52
草食家畜(2021年2期)2021-04-21 15:09:48
理化检验-化学分册(2021年4期)2021-04-18 14:41:50
中成药(2017年7期)2017-11-22 07:33:25
中成药(2017年5期)2017-06-13 13:01:12
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
中成药(2016年8期)2016-05-17 06:08:12
合成化学(2015年1期)2016-01-17 08:59:30
