
杂多酸/离子液体杂化材料催化苯的高选择性羟基化
2016-12-29 08:20:24童金辉宿玲弟王文慧马文梅薄丽丽
物理化学学报 2016年12期
马 青 童金辉,* 宿玲弟 王文慧 马文梅 薄丽丽
(1西北师范大学化学化工学院,兰州730070;2生态环境相关高分子材料教育部重点实验室,兰州730070;3甘肃农业大学理学院,兰州730070)
杂多酸/离子液体杂化材料催化苯的高选择性羟基化
马 青1,2童金辉1,2,*宿玲弟1,2王文慧1,2马文梅1,2薄丽丽3,*
(1西北师范大学化学化工学院,兰州730070;2生态环境相关高分子材料教育部重点实验室,兰州730070;3甘肃农业大学理学院,兰州730070)
制备了V取代的磷钼酸H3+xPMo12-xVxO40(x=0,1,2)及1-丁基-3-甲基咪唑溴盐离子液体([C4mim]Br),并采用离子交换的方法制备了系列杂化材料([C4mim]3+xPMo12-xVxO40,x=0,1,2);采用X射线衍射(XRD)、傅里叶变换红外光谱(FT-IR)、紫外-可见漫反射光谱(UV-Vis DRS)对所制备样品进行了表征;以H2O2为氧化剂,考察了所得样品催化苯羟基化制苯酚的活性。结果表明,和相应的离子液体及杂多酸相比,杂化材料的催化活性得到了很大的提高,尤其是催化剂[C4mim]5PMo10V2O40,在优化后的条件下,苯的转化率可达到21%,苯酚的选择性在99%以上。而且,该催化剂具有很好的可重复使用性,连续使用五次后,苯的转化率和苯酚的选择性没有明显降低。
杂多酸;离子液体;杂化材料;苯羟基化
1 引言
苯酚是石油化工、农用化学品和塑料等工业中使用最多的中间体之一,也是生产双酚A、尼龙-66和酚醛树脂等的必需化学品,市场需求量较大1,2。目前工业上生产的苯酚中超过95%是采用异丙苯氧化法3。这种方法需要加入多种有机试剂,并且副产物丙酮的市场需求量远小于苯酚,造成了严重的资源浪费和环境污染4。因此,在液相或气相中一步直接将苯羟基化制苯酚的方法成为研究的热点。在现有的报道中,常见的氧化剂有氮氧化物5、双氧水6和分子氧7等。其中以双氧水为氧化剂的方法,因操作简便、原子效率高,水为唯一的副产物而成为较为理想的方法。
杂多酸因具有独特的酸性、氧化还原性、结构和性质可调变性及在多种溶剂中的良好溶解性等特点,而被广泛用于催化液相氧化反应8,9。其中,钒掺杂的磷钼酸(PMoV)催化剂由于具有钒的氧化还原性、磷钼酸的氧化性和酸性的双重特性,而对氧化反应具有独特的催化活性10。研究表明,钒的取代度直接影响着PMoV催化剂的酸性、氧化还原性和热稳定性11,12。同时,含钒的催化剂已被证明是催化双氧水氧化苯羟基化反应的有效催化剂13,14。到目前为止,制备的各种含钒的催化剂如(C5H8O2)2VO15、[(CH3)4N]4PMo11VO4016和H5PMo10V2O40/SBA-1517等,在以双氧水为氧化剂,通过苯的羟基化制苯酚的反应中得到了较好的结果。然而,所报道的体系仍然面临着苯酚的选择性低、反应体系复杂、产物分离困难等问题。因此,发展高效、易分离的多相催化体系具有非常重要的意义。
通过将杂多酸催化剂负载到适当的载体上使其多相化的方法多有报道18-21。然而,在许多情况下,所得到的催化剂依然存在催化活性低、反应过程中活性物种易流失等问题20,21。目前,通过离子交换的方法制备的离子液体阳离子和杂多酸阴离子构成的杂化材料催化剂因具有较好的催化性能和可循环使用性能而引起了人们的广泛关注22-24。
本文采用离子交换法制备了一系列杂多酸/离子液体杂化材料催化剂[C4mim]3+xPMo12-xVxO40(x= 0,1,2),考察了杂多酸及其杂化材料对苯的羟基化反应的催化性能,对反应条件进行了优化,重点考察了反应温度、反应时间、催化剂用量和氧化剂用量等因素对反应结果的影响。同时,在最佳反应条件下考察了催化剂的可循环使用性能。
2 实验部分
2.1 实验仪器
晶相分析仪:日本岛津XRD-6000型X射线粉末衍射仪(Cu Kα射线);红外光谱仪:德国布鲁克vector-22型傅里叶红外光谱仪(分辨率2 cm-1);紫外-可见光谱仪:日本岛津UV-3100分光光度计;气质联用仪:美国惠普公司HP 6890/5973型气相色谱/质谱联用仪;气相色谱仪:美国Agilent 6890型气相色谱仪。
2.2 实验药品
所用试剂均为分析纯。1-甲基咪唑(C4H6N2)购于阿拉丁试剂有限公司;溴代正丁烷(C4H9Br),三氧化钼(MoO3),五氧化二钒(V2O5),磷酸(H3PO4),过氧化氢(H2O2,30%(w)),苯酚(C6H6O)均购于国药集团化学试剂有限公司。
2.3 实验方法
2.3.1 催化剂的制备
磷钼钒杂多酸(H3+xPMo12-xVxO40(x=0,1,2))是参照文献25的方法制备的。以H4PMo11VO40为例,具体的制备过程如下:分别称取15.8 g(0.11 mol) MoO3和0.91 g(0.005 mol)V2O5溶解于250 mL蒸馏水中。然后将上述混合液加热至373 K,并向其中逐滴加入85%(w)的磷酸1.15 g,保持15 h,悬浊液最终变为澄清透明的橙红色溶液;随后将溶液过滤,滤除其中未反应的杂质,将溶剂蒸干后得到橙红色的杂多酸H4PMo11VO40。其余两个样品的制备方法不变,只是所加入钒的含量不同。
离子液体[C4mim]Br是根据文献26中所描述的方法制备的。具体过程如下:将溴代正丁烷(13.7 g,0.1 mol)缓慢地加入1-甲基咪唑(8.2 g,0.1 mol)中,然后在40°C下搅拌12 h,得到一种浅黄色的粘稠液体。停止搅拌后加入适量的乙酸乙酯纯化,得到白色固体产物。
杂化材料[C4mim]3+xPMo12-xVxO40(x=0,1,2)是按照以下方法来制备的:根据[C4mim]Br与H3+xPMo12-xVxO40的不同摩尔比,先将[C4mim]Br溶解在30 mL去离子水中,然后将20 mL H3+xPMo12-xVxO40的水溶液在室温搅拌下逐滴加入,很快有橘黄色的沉淀生成。搅拌24 h使其充分反应后,过滤、洗涤,343 K下真空干燥24 h得杂化材料催化剂。
2.3.2 苯的羟基化
苯的羟基化反应是在装有回流冷凝管的25 mL圆底烧瓶中进行的。具体过程如下:先将1.0 mL (10 mmol)苯,5.0 mL乙腈和30 mg催化剂依次加入圆底烧瓶中,再将圆底烧瓶放入油浴中并加热至设定温度,然后在大约10 min内逐滴加入H2O2(30 mmol)后开始计时。反应结束后,离心分离出催化剂。产物通过HP6890/5973 GC/MS进行定性分析,以甲苯为内标,用Agilent 6890气相色谱进行定量分析。
3 结果与讨论
3.1 催化剂表征
3.1.1 FT-IR表征
所得样品的FT-IR谱图如图1所示。从图中可以看出所得杂多酸样品在1100-700 cm-1范围内出现了Keggin型结构杂多酸的四个明显的特征吸收峰(a,d,e):780-800 cm-1(Mo―Oc―Mo),860-880 cm-1(Mo―Ob―Mo),960-990 cm-1(Mo=Od)和1060-1080 cm-1(P―Oa)27,28。离子液体[C4mim]Br的FT-IR谱图(b)在3143和2955 cm-1处出现了C―H键的特征峰,在1566和1453 cm-1处出现了咪唑环上的C―N键的特征吸收峰。杂化材料的FT-IR谱图(c,f,g)中也出现了杂多酸的特征峰,表明在杂化材料中杂多酸依然保持了原来的结构。同时,和单一的杂多酸相比,杂化材料中杂多酸的特征峰发生了一定的位移,并且在1161和1055 cm-1处出现了P―O键的裂峰,表明离子液体阳离子和杂多酸阴离子之间有强的相互作用。同时,这些结果也初步证明了离子液体阳离子和杂多酸阴离子之间形成了离子键29。

图1 样品的FT-IR谱图Fig.1 FT-IR spectra of the samples
3.1.2 紫外-可见漫反射光谱(UV-Vis DRS)表征
所得样品的UV-Vis DRS谱图如图2所示。从图中可以看出,所有样品均在200-400 nm范围内有吸收峰。其中在206 nm附近的是O→P键的吸收峰,而在301 nm附近出现的较宽的吸收峰归属于Keggin单元中O2-→Mo6+之间的电荷迁移22。与单一的杂多酸相比,在杂化材料中206 nm处的吸收峰发生了蓝移。这表明离子液体和杂化材料之间存在一定的相互作用。

图2 样品的UV-Vis漫反射谱图Fig.2 UV-Vis DRS of the samples
3.1.3 XRD表征
图3是所制备样品的XRD谱图。在2θ为8°和27°处所有的杂多酸样品均出现了杂多酸晶体的明显的特征衍射峰30,31。然而,对于杂化材料,27°的峰变弱甚至消失,而8°左右的衍射峰有明显的增强,同时在17°和22°处出现了新的衍射峰。这个结果表明Keggin型阴离子和离子液体阳离子之间可能形成了三维晶体结构32。

图3 样品的XRD衍射谱图Fig.3 XRD patterns of the samples

表1 不同催化剂催化苯羟基化性能比较aTable 1 Catalytic behavior of different catalysts on the hydroxylation reaction of benzenea
3.2 催化剂催化性能的考察
以H2O2为氧化剂,乙腈为溶剂,考察了不同催化剂催化苯的羟基化反应的性能,结果如表1所示。从表中可以看出,在相同的反应条件下,当不加催化剂(Entry 1),或以离子液体[C4mim]Br (Entry 2),杂多酸H3PMo12O40(Entry 3)及其杂化材料[C4mim]3PMo12O40(Entry 6)为催化剂时,苯不能被氧化。然而,当以V取代的杂多酸H4PMo11VO40和H5PMo10V2O40(Entries 4,5)为催化剂时,苯的转化率分别达到了11%和12%。这和文献报道的结果一致:杂多酸中的V离子是苯羟基化反应的活性中心,反应过程中V(5+)-OO·离子被还原成了V (4+)-OO·13,33。而当以杂化材料[C4mim]4PMo11VO40和[C4mim]5PMo10V2O40为催化剂时,苯的转化率分别为17%和21%(Entries 7,8),和相应的杂多酸相比,杂化材料对苯的羟基化反应的转化率分别提高了6%和9%。同时,苯酚的选择性都在99%以上。这一结果表明将杂多酸与离子液体杂化可以显著地提高其对苯的羟基化反应的催化活性。杂化材料的催化性能之所以能够得到显著提高,一个很重要的原因在于亲水的杂多酸和疏水的离子液体杂化后,很好地平衡了所得杂化材料表面的亲-疏水性质,提高了苯在反应介质中的溶解性及与氧化剂H2O2的混合度。这一点在Chen等34的研究中也得到过证实。因此,以下选择了[C4mim]5PMo10V2O40材料作为催化剂,对苯羟基化反应的条件进行了优化。
3.2.1 不同催化剂用量的影响
我们考察了不同催化剂用量对苯的羟基化反应的影响,结果如图4所示。从图中可以看出,随着催化剂用量从15 mg增加到30 mg,苯的转化率也从12%增加到了21%,而当进一步增加催化剂的用量至35 mg时,苯的转化率只增加了2%。苯酚的选择性则不受催化剂用量的影响,始终保持在99%以上。因此,在后续的反应中,选择催化剂的用量为30 mg。
3.2.2 不同氧化剂用量的影响
双氧水用量对苯的羟基化反应的影响如图5所示。从图中可以看出,当H2O2用量从10 mmol (30%H2O2,1 mL)增加到30 mmol(30%H2O2,3 mL)时,苯的转化率从9%增加到了21%,苯酚的选择性保持在99%以上。当进一步增加H2O2的用量时,苯的转化率和苯酚的选择性均基本保持不变。因此,在后续的反应中,选择H2O2的用量为30 mmol。

图4 不同催化剂用量对苯的羟基化反应的影响Fig.4 Effect of catalyst amount on the hydroxylation of benzene

图5 不同氧化剂用量对苯的羟基化反应的影响Fig.5 Effect of oxidant amount on the hydroxylation of benzene
3.2.3 不同反应温度的影响
不同反应温度对苯的羟基化反应的影响如图6所示。从图中可以看出,当反应温度从50°C升高到65°C时,苯的转化率从11%增加到了16%,当进一步升高反应温度到70°C时,苯的转化率增加到了21%,同时苯酚的选择性一直保持在99%以上。因此,在后续的反应中,选择70°C作为反应温度。
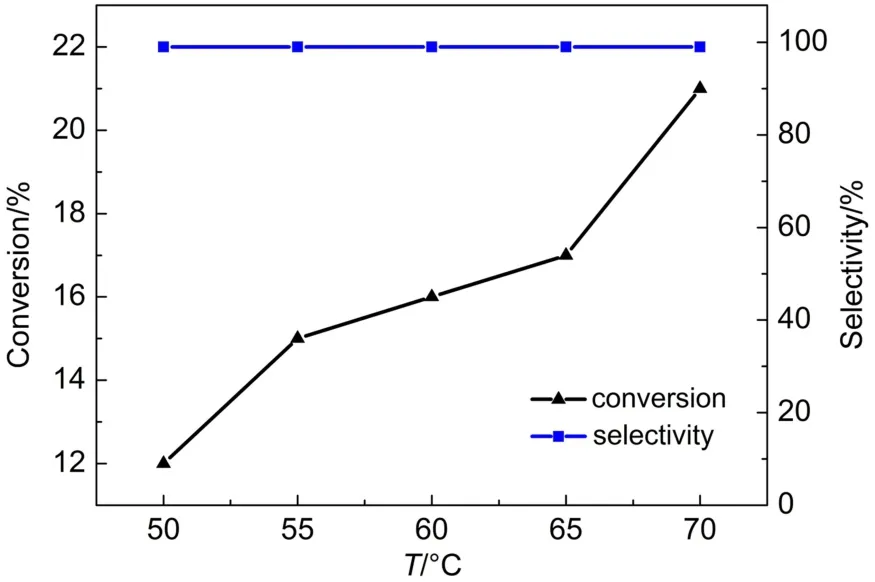
图6 不同反应温度对苯的羟基化反应的影响Fig.6 Effect of reaction temperature on the hydroxylation of benzene
3.2.4 不同反应时间对催化性能的影响
不同反应时间对苯的羟基化反应的影响如图7所示。从图中可以看出,当反应时间从3 h增加到6 h时,苯的转化率从12%增加到了21%,苯酚的选择性基本保持不变。当进一步延长反应时间至7 h时,苯的转化率只增加了2%,而苯酚的选择性均在99%以上。反应5 h以后,苯的转化速率明显降低,这主要是由于H2O2的分解导致其浓度降低造成的。从图7可以看出,当反应时间从3 h延长至5 h时,H2O2的利用率从29%上升到了41%;当反应时间进一步延长至7 h时,H2O2的利用率则从41%下降到了32%。从催化效率的角度考虑,选择6 h作为本实验条件下的反应时间。
3.2.5 催化剂的循环使用性能
反应结束后,离心分离出催化剂,80°C下真空干燥,然后在相同条件下重复使用。结果如图8所示,循环反应5次之后,苯酚的选择性仍然保持在99%以上,但是苯的转化率却从21%降到了19%。苯的转化率的降低主要归因于回收过程中催化剂不可避免的流失。循环5次的过程中,苯的平均转化率为20%。这一结果表明催化剂[C4mim]5PMo10V2O40具有较好的稳定性和可循环使用性能。

图7 不同反应时间对苯的羟基化反应的影响Fig.7 Effect of reaction time on the hydroxylation of benzene
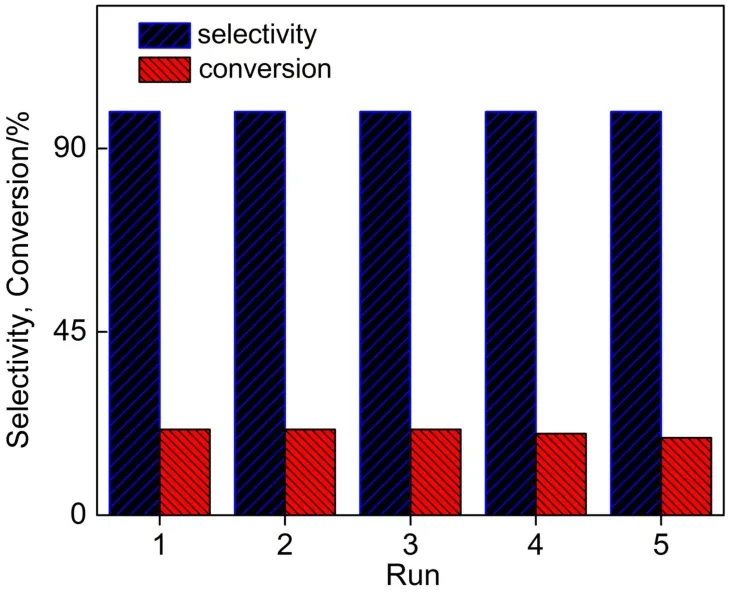
图8 催化剂[C4mim]5PMo10V2O40的重复使用Fig.8 Reusability of the catalyst[C4mim]5PMo10V2O40
为了进一步考察催化剂的稳定性和多相性,我们进行了中断反应实验:在反应进行到4 h后,滤出催化剂,将反应液继续投入反应器进行反应至6 h,三次实验所得苯的平均转化率为14.8%。另外,考虑到反应过程中H2O2的分解,在中断反应过程中,在滤出催化剂后,将反应液继续投入反应器,并向其中补加0.45 mL(为原加入量的15%)的H2O2(30%(w)),继续反应至6 h,三次实验所得苯的平均转化率为15.2%。无论补加还是不补加H2O2,苯酚的选择性均在99%以上。上述中断反应结果和一次6 h连续反应至4 h的结果一致,说明该催化剂具有良好的稳定性和多相性。
4 结论
采用简单的离子交换方法制备了系列杂多酸/离子液体杂化材料催化剂([C4mim]3+xPMo12-xVxO40,x=0,1,2),通过考察所制备催化剂催化H2O2氧化苯制苯酚的催化活性发现,只有V取代的杂多酸和杂化材料能够催化苯的羟基化反应,且其催化活性随V取代量的增加而增加。而且,和相应的杂多酸相比,杂化材料的催化活性得到了很大提高;更重要的是,在我们所考察的条件下,苯酚的选择性均在99%以上。尤其是杂化材料催化剂[C4mim]5PMo10V2O40,在优化了的反应条件下,重复使用5次之后,苯的平均转化率达到了20%,且产物苯酚的选择性保持在99%以上。这一结果不仅为杂多酸/离子液体杂化材料的活性增强提供了一个很好的例证,同时为苯的羟基化建立了一个高效、高选择性、较为绿色的催化体系。
(1) Zhang,J.;Tang,Y.;Li,G.;Hu,C.Appl.Catal.A 2005,278, 251.doi:10.1016/j.apcata.2004.10.009
(2) Wang,M.;Leitch,M.;Xu,C.C.J.Ind.&Eng.Chem.2009,15, 870.doi:10.1016/j.jiec.2009.a)09.015
(3) Zheng,Z.H.;Guo,W.L.;Fan,J.G.Petrochem.Technol.2004, 33,1096.[郑朝晖,郭卫玲,范金钢.石油化工,2004,33,1096.]
(4) Sumimoto,S.;Tanaka,C.;Yamaguchi,S.T.;Ichihashi,Y.; Nishiyama,S.;Tsuruya,S.Ind.Eng.Chem.Res.2006,45,7444. doi:10.1021/ie060743e
(5) Molinari,R.;Poerio,T.;Argurio,P.Catal.Today 2006,118,52. doi:10.1016/j.cattod.2005.11.089
(6) Yuan,C.;Gao,X.;Pan,Z.;Li,X.;Tan,Z.Catal.Commun. 2015,58,215.doi:10.1016/j.catcom.2014.08.004
(7) Chen,L.H.;Li,W.H.;Li,D.;Han,F.Chem.Ind.&Eng.Pro. 2005,24,236.[陈练洪,李稳宏,李 冬,韩 枫.化工进展, 2005,24,236.]
(8) Hajian,R.;Tangestaninejad,S.;Moghadam,M.;Mirkhani,V.; Mohammadpoor-Baltork,I.;Khosropour,A.R.J.Coord.Chem. 2011,64,4134.doi:10.1080/00958972.2011.636038
(9) Gu,L.Y.;Gao,B.J.;Fang,X.L.Acta Phys.-Chim.Sin.2013, 29,191.[顾来沅,高保娇,房晓琳.物理化学学报,2013,29, 191.]doi:10.3866/PKU.WHXB201210266
(10) Rao,P.;Rao,K.V.;Prasad,P.S.;Lingaiah,N.Catal.Commun. 2010,11,547.doi:10.1016/j.catcom.2009.12.016
(11) Boudjema,S.;Vispe,E.;Choukchou-Braham,A.;Mayoral,J. A.;Bachir,R.;Fraile,J.M.RSC Adv.2015,5,6853. doi:10.1039/c4ra13604g
(12) Yuan,C.;Gao,X.;Pan,Z.;Li,X.;Tan,Z.Catal.Commun. 2015,58,215.doi:10.1016/j.catcom.2014.08.004
(13) Zhang,F.;Maiping,G.;Hanqingd,G.;Jun,W.Chin.J.Chem. Eng.2007,15,895.doi:10.1016/S1004-9541(08)60021-X
(14) Antonyraj,C.A.;Srinivasan,K.Catal.Surv.Asia 2013,17,47. doi:10.1007/s10563-013-9153-8
(15) Molinari,R.;Lavorato,C.;Poerio,T.Appl.Catal.A 2012,417, 87.doi:10.1016/j.apcata.2011.12.031
(16) Chen,J.;Li,J.;Zhang,Y.;Gao,S.Res.Chem.Intermed.2010, 36,959.doi:10.1007/s11164-010-0208-4
(17) Kharat,A.N.;Moosavikia,S.;Jahromi,B.T.;Badiei,A.J. Mol.Catal.A:Chem.2011,348,14.doi:10.1016/j. molcata.2011.07.014
(18) Kholdeeva,O.;Maksimchuk,N.;Maksimov,G.Catal.Today 2010,157,107.doi:10.1016/j.cattod.2009.12.016
(19) Zhao,P.;Leng,Y.;Zhang,M.;Wang,J.;Wu,Y.;Huang,J. Chem.Commun.2012,48,5721.doi:10.1039/c2cc31919e
(20) Wang,X.X.;Xu,H.L.;Shen,W.;Ruhlmann,L.;Qin,F.;Sorgues,S.;Colbeau-Justin,C.Acta Phys.-Chim.Sin.2013,29, 1837.[王晓夏,徐华龙,沈 伟,Ruhlmann,L.秦 枫, Sorgues,S.,Colbeau-Justin,C.物理化学学报,2013,29,1837.] doi:10.3866/PKU.WHXB201307024
(21) Rezvani,M.A.;Oveisi,M.;Asli,M.A.N.J.Mol.Catal.A: Chem.2015,410,121.doi:10.1016/j.molcata.2015.09.010
(22) Liang,R.;Chen,R.;Jing,F.;Qin,N.;Wu,L.Dalton Trans. 2015,44,18227.doi:10.1039/c5dt02986d
(23) Zhu,W.;Huang,W.;Li,H.;Zhang,M.;Jiang,W.;Chen,G.; Han,C.Fuel Process.Technol.2011,92,1842.doi:10.1016/j. fuproc.2011.04.030
(24) Zhang,M.;Li,M.;Chen,Q.;Zhu,W.;Li,H.;Yin,S.;Li,Y.;Li, H.RSC Adv.2015,5,76048.doi:10.1039/c5ra13787j
(25) Jing,L.;Shi,J.;Zhang,F.;Zhong,Y.;Zhu.W.J.Ind.Eng. Chem.Res.2013,52,10095.doi:10.1021/ie4007112
(26) Burrell,A.K.;Del Sesto,R.E.;Baker,S.N.;McCleskey,T.M.; Baker,G.A.Green Chem.2007,9,449.doi:10.1039/b615950h
(27) Tsigdinos,G.A.;Hallada,C.J.Inorg.Chem.1968,7,437. doi:10.1021/ic50061a009
(28) Benaissa,H.;Davey,P.;Khimyak,Y.;Kozhevnikov,I.J.Catal. 2008,253,244.doi:10.1016/j.jcat.2007.11.011
(29) Zhao,P.;Wang,J.;Chen,G.;Zhou,Y.;Huang,J.Catal.Sci. Technol.2013,3,1394.doi:10.1039/c3cy20796j
(30) Xue,S.;Chen,G.;Long,Z.;Zhou,Y.;Wang,J.RSC Adv.2015, 5,19306.doi:10.1039/c4ra15921g
(31) Wang,R.;Jia,D.;Cao,Y.Electrochim.Acta 2012,72,101. doi:10.1016/j.electacta.2012.04.011
(32) Zhao,P.;Zhang,M.;Wu,Y.;Wang,J.Ind.Eng.Chem.Res. 2012,51,6641.doi:10.1021/ie202232j
(33) Raj,N.K.K.;Ramaswamy,A.V.;Manikandan,P.J.Mol. Catal.A:Chem.2005,227,37.doi:10.1016/j. molcata.2004.10.005
(34) Chen,G.J.;Zhou,Y.;Long,Z.T.;Wang,X.C.;Li,J.;Wang,J. ACS Appl.Mater.Inter.2014,4438.doi:10.1021/am5001757
Highly Selective Hydroxylation of Benzene Catalyzed by Hybrids of Polyoxometalate/Ionic Liquid
MAQing1,2TONG Jin-Hui1,2,*SU Ling-Di1,2WANG Wen-Hui1,2MAWen-Mei1,2BO Li-Li3,*
(1College of Chemistry and Chemical Engineering,Northwest Normal University,Lanzhou 730070,P.R.China;2Key Laboratory of Eco-Environment-Related Polymer Materials,Ministry of Education,Lanzhou 730070,P.R.China;3College of Science,Gansu Agricultural University,Lanzhou 730070,P.R.China)
Aseriesofhybridmaterials([C4mim]3+xPMo12-xVxO40,x=0,1,2)basedonV-substitutedphosphomolybdic acidH3+xPMo12-xVxO40(x=0,1,2)and ionic liquid 1-butyl-3-methyl imidazolium bromide([C4mim]Br)have been prepared by an anion-exchange method.The samples were characterized by X-ray diffraction(XRD),Fourier transform infrared spectrophotometry(FT-IR)and UV-Vis diffuse reflectance spectra(UV-Vis DRS)analysis. The catalytic performances of the samples were tested in oxidation of benzene to produce phenol using H2O2as the oxidant.The results showed that the hybrids[C4mim]3+xPMo12-xVxO40exhibit much higher catalytic properties than both the corresponding moieties.In particular,under the optimized conditions,21%of benzene conversion and 99%selectivity for phenol have been obtained with[C4mim]5PMo10V2O40.The sample also exhibits good reusability and was reused five times without a significant decrease in conversion and selectivity.
Polyoxometalate;Ionic liquid;Hybrid material;Hydroxylation of benzene
O643
10.3866/PKU.WHXB201609181
Received:July 25,2016;Revised:September 16,2016;Published online:September 18,2016.
*Corresponding author.Email:jinhuitong@126.com;Tel:+86-931-62751724.
The project was supported by the National Natural Science Foundation of China(51302222,21363021,21202133),Program for Changjiang Scholars and Innovative Research Team in University,China(IRT15R56),Program for UndergraduateAcademic Innovative Research Team of Northwest Normal University,China.
国家自然科学基金(51302222,21363021,21202133),教育部长江学者和创新团队发展计划(IRT15R56)和西北师范大学本科生学术能力提升计划项目资助
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:54
陕西中医(2018年6期)2018-08-29 00:43:34
中成药(2018年2期)2018-05-09 07:20:05
新乡学院学报(2016年6期)2016-12-01 05:21:38
中国塑料(2016年1期)2016-05-17 06:13:00
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
当代化工研究(2016年9期)2016-03-20 16:22:11
合成化学(2015年4期)2016-01-17 09:01:27
化工生产与技术(2014年6期)2014-02-27 13:42:07
