
In-Au(111)和Ir-Au(111)合金表面的性质及其对巴豆醛的吸附比较
2016-12-29 08:20:16蒋军辉钱梦丹薛继龙夏盛杰倪哲明邵蒙蒙
物理化学学报 2016年12期
蒋军辉 钱梦丹 薛继龙 夏盛杰 倪哲明 邵蒙蒙
(浙江工业大学化学工程学院,杭州310014)
In-Au(111)和Ir-Au(111)合金表面的性质及其对巴豆醛的吸附比较
蒋军辉 钱梦丹 薛继龙 夏盛杰 倪哲明*邵蒙蒙
(浙江工业大学化学工程学院,杭州310014)
采用密度泛函理论研究了M(M=In,Ir)原子修饰的M-Au(111)合金表面的稳定性,并选其最优模型探讨了合金表面的活性及其对巴豆醛的吸附。合金的几何构型、形成能和结合能等性质表明,In-Au(111)面的稳定性随In原子的间距增大而提高,Ir-Au(111)面的稳定性随Ir原子的间距增大而降低。对于巴豆醛在MAu(111)面上的吸附,当其通过C=O吸附于合金表面的TopM位时,吸附能最大,吸附构型最稳定。从巴豆醛的结构变化、态密度、差分电荷密度以及Mulliken电荷布居等分析可以看出,稳定吸附构型的巴豆醛分子形变较大,电荷转移明显。其中,位于-7.04 eV至费米能级处的p、d轨道杂化,对体系的吸附具有重要贡献。分析比较In-Au(111)面与Ir-Au(111)面,发现后者的配体效应更佳,不仅具有更高的稳定性和活性,而且对于巴豆醛具有更强的吸附力。此外,相比于改性前的Au(111)面,M原子的修饰明显提升了金属表面的稳定性及吸附能力。
In-Au(111)面;Ir-Au(111)面;修饰;巴豆醛;吸附;密度泛函理论
1 引言
1987年,Haruta等1发现负载在氧化物上的纳米Au对低温下的CO氧化反应具有很好的催化活性,自此打破了Au催化惰性的观念,打开了Au催化应用的新篇章。Bus等2研究了Au/Al2O3以及Pt/ Al2O3催化剂对肉桂醛的催化加氢反应,发现Au/ Al2O3的催化活性明显高于Pt/Al2O3。Bailie和Hutching3通过Au/ZrO2催化剂首次完成了巴豆醛的C=O选择性氢化。蒋军辉等4探讨了巴豆醛在Au (111)上的选择性催化加氢机理,结果表明Au(111)面对于巴豆醛的C=O具有较好的选择性氢化能力。虽然纯Au催化剂具有较好的低温氧化能力以及较高的选择性,但是纯Au催化剂存在活性低且易烧结等缺陷5,6。因此,对于Au催化剂的改良,将有助于其未来的发展和应用范围的开阔。
目前,实验和理论研究上改善单金属催化剂的方法有载体和前驱体的替换7、催化剂活性粒子的改变以及第二元素的修饰8,9。其中第二元素的修饰可优化催化剂的局域电子效应和几何构型,生成双金属混合位,从而增强催化剂的活性,进而提高其催化性能。张连阳等10探讨了Au修饰的Au/ Pd(111)面的稳定性,同时研究了噻吩在其表面的吸附和加氢脱硫机理,发现改性后的催化剂具有更高的稳定性以及催化活性。Cárdenas-Lizana等11研究了Au/Al2O3以及PdAu/Al2O3催化剂在氯代硝基苯加氢中的催化性能,发现Au/Al2O3催化剂的选择性较好,但活性较低。Pd修饰后的PdAu/Al2O3催化剂的催化活性明显提高,且对氯代硝基苯的选择性为100%。
巴豆醛(CAL)是α,β-不饱和醛的典型代表之一,其一系列加氢产物作为重要的化工原料在精细化学品的合成当中具有广泛应用12-14。例如,C=O选择性氢化得到的巴豆醇是医药及香精的中间体,C=C选择性氢化得到的丁醛是树脂以及杀虫剂的中间体。近年来,第二元素修饰的Au催化剂被普遍应用于α,β-不饱和醛的加氢反应中。Yang等15探究了SBA-15负载的Au和In-Au对于CAL的催化加氢,结果显示In-Au/SBA-15催化剂的C=O选择性催化能力更强,不仅对于巴豆醇的选择性和产率分别高达78%和71%,而且催化剂的循环使用并不影响其选择性。He等16计算表明,In对于Au(110)面的修饰,显著增强了其与丙烯醛的相互作用,同时也提高了对于丙烯醇的选择性。Rojas等17分析了Ir-Au/TiO2的性质及其对于柠檬醛的催化加氢,一系列的表征表明Ir-Au/TiO2催化剂具有较好的稳定性,并且相对于Au/TiO2,合金在柠檬醛的选择性催化加氢中具有更高的催化活性。Zhao等18研究表明,对于肉桂醛的催化加氢反应,Ir修饰的Ir-Au催化剂相比较于Au催化剂,既有较高的C=O选择性,又有较强的H2活化能力。
综上,在Au催化剂中掺杂M(M=In,Ir)原子所形成的修饰M-Au合金,具有优异的α,β-不饱和醛选择性催化加氢性能。然而,有关M-Au合金的性质及其对于CAL吸附的理论研究较少。因此,本研究采用密度泛函理论(DFT)构建了M原子修饰的M-Au(111)面,对可能的合金模型的稳定性和活性进行了分析,同时研究了CAL在M-Au(111)面上的吸附行为及其优势吸附构型的电子性质,以期为M-Au合金催化剂的设计及CAL在其表面的选择性加氢提供理论依据。
2 计算方法和模型
本研究的DFT计算利用Materials Studio 5.5软件包中的DMol3程序包进行19,20。为了减小范德华力引起的吸附误差,本研究运用了DFT-D的方法。电子交换相关泛函基于广义梯度积分(GGA)的Perdew-Wang-91(PW91)泛函21,价电子波函数采用双数值极化函数(DNP)展开,且未限制电子自旋。计算精度设为medium,smearing设为13.10 kJ·mol-1,k点设为3×3×1。结构优化合理性的判据为:能量差异小于4.80×10-2kJ·mol-1,原子位移小于5.00×10-4nm以及各个原子上的力小于9.70 kJ·mol-1·nm-1。
本文首先选用低覆盖率(1/16)p(4×4)的周期性四层平板模型来模拟Au(111)面,且将真空层厚度选为1.20 nm,避免了平板间发生镜像作用。在此基础上,通过2个修饰原子取代Au(111)表面的2个Au原子后构成M-Au(111)面18,22,并根据修饰原子取代位置的相邻距离不同,构建了如图1所示由近及远的3种模型分别记为(a)、(b)、(c)。计算过程中考虑到表面弛豫的影响,对底下2层原子进行了固定,上2层原子可自由移动。图1同时列出了CAL的结构。
3 结果与讨论
3.1 M-Au(111)面性质分析

图1 M-Au(111)面的不同模型和CAL的结构Fig.1 Different models of M-Au(111)surfaces and the structure of CAL
3.1.1 M-Au(111)面稳定性分析
M-Au(111)面是以Au为溶剂,在其晶格点阵中置换溶入溶质原子M所形成的置换合金表面,其中原子尺寸因素(ΔR)大小是影响其形成的关键23。现根据计算模型将ΔR定义为:ΔR=|RMRAu|/RAu,式中:RM为溶质原子M的原子半径(RIn= 0.166 nm,RIr=0.136 nm),RAu为溶剂原子Au的原子半径(RAu=0.146 nm)。当ΔR<0.150时,溶质M与溶剂Au之间可以形成稳定的合金,且ΔR越小,引起的晶格畸变越小。表1列出了M-Au(111)面的ΔR,由表1可见,对于In-Au(111)面和Ir-Au(111)面,其ΔR都小于0.150,分别为0.137和0.068。因此,In和Ir原子都能够较好地与Au面形成合金,并且Ir原子置换所引起的晶格畸变较In原子小。从而可以推测,Ir-Au(111)面较In-Au(111)面的稳定性更高。
表1同时列出了优化后合金的M―Au键平均键长d以及置换原子M在M-Au(111)面上的凸起高度h,进而从几何构型角度展现了模型的变化情况。在In-Au合金中,模型(a)、(b)、(c)的In―Au键平均键长分别为0.300、0.299以及0.297 nm,并且In原子分别凸出表面0.092、0.091以及0.089 nm。这可能是由于RIn大于RAu,以及Au、In原子晶体结构的差异(Au属于立方晶系,In属于四方晶系),使得In―Au键平均键长大于Au―Au键(0.288 nm),从而引起修饰表面凸起。同时由于模型(c)的几何形变最小,因此可以推测In以(c)模型修饰Au(111)面的可能性最大。在Ir-Au合金中,模型(a)、(b)、(c)的Ir―Au键平均键长分别为0.283、0.287、0.287 nm,并且Ir原子较表面分别下凹0.036、0.033、0.033 nm。引起表面下凹的可能原因有两个,其一是RIr小于RAu,其二是Ir和Au之间的电负性差值大于Au和Au之间的电负性差值,引起的Ir―Au的作用力大于Au―Au的作用力。对比分析三种模型可得,在Ir-Au(111)面中模型(a)的稳定性最强,模型(c)的稳定性最弱。综上,相比于In原子,Ir原子的结构性质更加接近Au原子,从而使得Ir-Au(111)面的稳定性较In-Au(111)面的稳定性更高。
为了进一步研究M-Au合金的稳定性,本文还探讨了合金的形成能(ΔEf)和结合能(ΔEc)等热力学性质。其中ΔEf定义为:ΔEf=EAu(111)+2EM-(EM-Au(111)+2EAu),式中EM-Au(111)和EAu(111)分别为M-Au (111)面和Au(111)面的能量,EAu和EM分别为孤立Au和M原子的能量。ΔEc定义为:ΔEc=(∑Ei-EM-Au(111)/Au(111))/64,式中Ei为孤立原子(Au,In,Ir)的能量,EM-Au(111)/Au(111)为M-Au(111)面或Au(111)面的能量。表1显示In-Au(111)面不同模型的形成能随着表面In原子间距的增加近似递增,而对于Ir-Au (111)面恰好相反。根据形成能的定义,形成能越大,体系越稳定。In-Au(111)面的模型稳定性顺序为(c)>(b)>(a),Ir-Au(111)面的模型稳定性顺序为(a)>(b)>(c),且Ir-Au(111)面的稳定性高于In-Au (111)面。对于结合能,In-Au(111)面和Ir-Au(111)面的模型(a)、(b)、(c)能量分别为 391.15、391.54、391.71以及425.78、425.52、425.44 kJ·mol-1,且都较Au(111)面的能量(265.66 kJ·mol-1)大。根据结合能定义,结合能是由原子自由态形成化合物所释放的能量。因此,Ir-Au(111)面的稳定性高于In-Au(111)面,同时都比Au(111)面的稳定性要好。In-Au(111)面的构型稳定性顺序为(c)>(b)>(a),Ir-Au(111)面的构型稳定性顺序为(a)>(b)>(c),此结论与形成能以及几何结构分析结果一致。

表1 M-Au(111)面不同模型的平均凸起高度、M―Au键平均键长、形成能以及结合能Table 1 Geometrical parameters of the M-Au(111)surfaces,including the average embossment height of doped atoms, average bond length of M―Au,formation and cohesive energies
综上所述,In原子和Ir原子都能够较好地通过置换取代的方法与Au(111)面形成相应的稳定合金,并且Ir-Au(111)面的稳定性高于In-Au(111)面。In-Au(111)面和Ir-Au(111)面上的最稳定模型分别为(c)和(a),因此这两类模型存在的可能性最大。下文研究将以In-Au(111)面的模型(c)以及Ir-Au(111)面的模型(a)为基础,对其进行化学活性以及吸附性能的探讨。图2给出了其优化后的M-Au (111)面平板模型。
3.1.2 M-Au(111)面化学活性分析
d带中心可以较好地反映出金属表面的化学活性,且其与费米能级的间距越小,化学活性越高24。对于M-Au(111)面,d带电子对总态密度的贡献最大。第二金属的修饰使得合金表面的电荷密度发生变化,从而导致d带电子的变化,进而可用d带中心的大小来预测和验证不同金属表面的吸附能力。计算表明,In-Au(111)面和Ir-Au(111)面的d带中心分别位于-3.54和-3.48 eV,都比Au(111)面(-3.63 eV)更靠近费米能级。因此In和Ir原子对Au(111)面的修饰,使得合金的d带中心向费米能级迁移,并且Ir-Au(111)面的d带中心向右迁移的距离较In-Au(111)面更远,这可能是由于Ir和Au原子间的相互作用大于In原子,即Ir原子修饰引起的配体效应大于In原子。据此可以推测金属表面吸附能力的大小为Ir-Au(111)面>In-Au(111)面>Au(111)面。另外,Mulliken电荷布居也是一种基于轨道分布定量描述体系电荷分布情况的分析方法25。In和Ir原子加入后,金属表面上Au原子的电荷由-0.01e分别降低为-0.02e和-0.06e,而In和Ir原子上的电荷分别增加到0.18e和0.47e。所以可以推断,在M-Au(111)面上,吸附将围绕M原子展开,且在Ir原子上的吸附力大于In原子,此结论与d带中心预测结果一致。
3.2 M-Au(111)面上CAL的吸附
3.2.1 吸附构型及其吸附能
CAL催化加氢过程首先是CAL吸附于催化剂M-Au(111)面,进而CAL与解离的H原子发生选择性氢化反应,最终得到的产物脱离M-Au(111)表面,因此研究CAL在合金催化剂表面的吸附是探究催化加氢的关键步骤。根据本课题组之前的研究以及相关文献报道4,26,CAL存在单点(O,C=O及C=C)和双点(O协同C=C或C=O协同C=C)两种吸附点位。在In-Au(111)面上,CAL可通过单点吸附于TopAu、TopIn、BriAu-Au、BriAu-In、Hcp以及Fcc六种吸附位置,共计18种吸附构型,通过双点吸附于上述六种吸附位置,共计36种吸附构型,总计优化了54种吸附构型。在Ir-Au(111)面上,CAL可通过单点吸附于 TopAu、TopIr、BriAu-Au、BriAu-Ir、BriIr-Ir、Hcp以及Fcc七种吸附位置,共计21种吸附构型,通过双点吸附于上述七种吸附位置,共计49种吸附构型,总计优化了70种吸附构型。为了确定其吸附的程度,本研究引入了吸附能(Eads)一值,并定义为:Eads=ECAL+EM-Au(111)-ECAL/M-Au(111),式中ECAL/M-Au(111)表示CAL吸附在M-Au (111)面时体系的能量。Eads越大表示吸附体系越稳定。
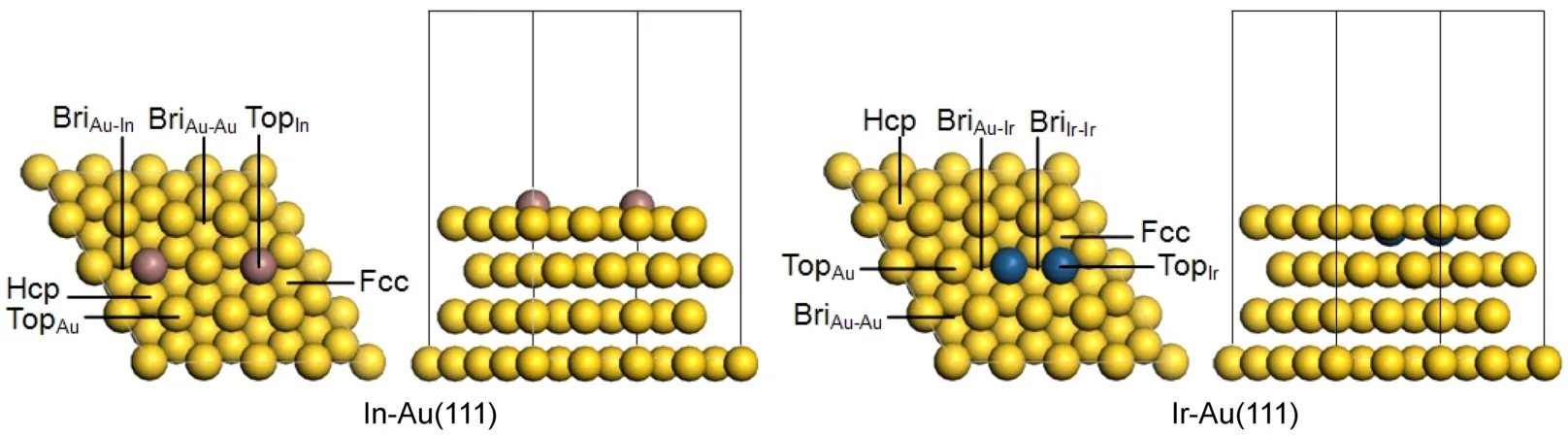
图2 M-Au(111)面稳定平板模型的俯视和侧视图Fig.2 Top and side views of the stable M-Au(111)plate models
表2和表3分别列出了优化后CAL在In-Au (111)面和Ir-Au(111)面上能够稳定存在的吸附构型及其吸附能值和O原子距离表面的高度(dO-layer)。优化后CAL在In-Au(111)面和Ir-Au(111)面上分别只存在17种和22种稳定吸附构型。引起吸附构型减少的可能原因是M原子的修饰使得M-Au(111)面对于CAL具有较大的吸附力,从而引起构型的普遍变化。同时,M-Au(111)面上不均匀分布的电荷对于吸附构型的减少也有一定影响。特别是对于双点吸附构型,分别缩减到9种和12种。这可能是由于甲基引起的空间阻碍效应较M-Au(111)面对于CAL的C=C吸附力更强,使得双点吸附较难存在。对于CAL/In-Au(111)体系,其吸附能在79.88-145.45 kJ·mol-1之间,O原子距离表面为0.242-0.595 nm之间。当CAL以C=O吸附于TopIn位时,即以η2-diσCO(TopIn)构型存在时,其吸附能最大,为145.45 kJ·mol-1,O原子距离表面为0.242 nm。且吸附前后,C=O以及C=C的键长分别被拉长0.003和0.002 nm,连接C=O和C=C的C―C键长缩短0.002 nm,说明该种吸附方式导致了分子的键长平均化。对于CAL/Ir-Au(111)体系,其吸附能在85.18-152.21 kJ·mol-1之间,O原子距离表面为0.173-0.471 nm之间。当CAL以C=O吸附于TopIr位时,即以η2-diσCO(TopIr)构型存在时,其吸附能最大,为152.21 kJ·mol-1,较Ir(111)面得到提高27,O原子距离表面为0.233 nm。且吸附前后,C=O以及C=C的键长分别被拉长0.004和0.002 nm,连接C=O和C=C的C―C键长缩短0.003 nm,同样说明该类吸附方式导致了分子的键长平均化。图3分别为CAL在In-Au(111)面和Ir-Au (111)面上的最稳定吸附构型。
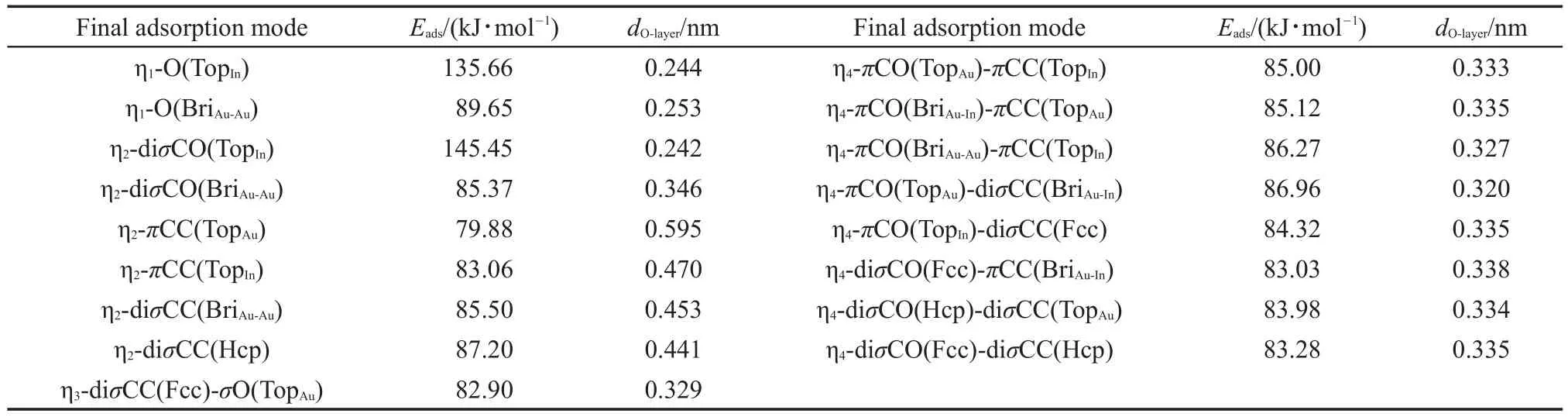
表2 CAL在In-Au(111)面上不同吸附位的计算结果Table 2 Calculation results of CALadsorption at different sites of In-Au(111)surface

表3 CAL在Ir-Au(111)面上不同吸附位的计算结果Table 3 Calculation results of CALadsorption at different sites of Ir-Au(111)surface

图3 CAL在M-Au(111)面上的最稳定吸附构型Fig.3 The most stable adsorption configurations of CALon M-Au(111)surfaces
综上,当CAL的C=O以σ键吸附于In-Au (111)面和Ir-Au(111)面的TopM位时最稳定,其对应的吸附能最大,表现为化学吸附。相比较该两种稳定吸附构型,对于吸附能,CAL在Ir-Au(111)面上的吸附能略高于In-Au(111)面,同时都高于Au (111)面(93.70 kJ·mol-1)。对于键长变化,CAL在Ir-Au(111)面上的键长变化较In-Au(111)面更加明显,且C=O的键长变化也相对较大,从而可以推测CAL的C=O最容易受到破坏。因此,Ir-Au (111)面对CAL的吸附作用比In-Au(111)面对CAL的吸附作用更强。此外,相对于Pt(111)面26,28、Sn/ Pt(111)面29和Au团簇30,M-Au(111)面表现出更强的CAL吸附能力,并且由于其独特的C=O吸附构型,使之更有利于C=O的活化。
3.2.2 吸附构型的电子性质分析
由上述分析可知,M原子的修饰改善了Au (111)面的吸附性能。为进一步了解CAL与M-Au (111)面的电子交互作用,本文对最稳定吸附构型的态密度进行了分析,如图4所示。对于CAL/MAu(111)体系,在-17.18至-7.04 eV处,CAL与M-Au(111)面的电子杂化作用较弱,但CAL分子内的s和p轨道杂化作用明显,因此可以推测其主要构键方式为共价键。在-7.04 eV至费米能级处,主要是M-Au(111)的d轨道与CAL的p轨道发生杂化作用,这是CAL能够稳定吸附于M-Au(111)面的主要原因,M-Au(111)面的d轨道与CAL的s轨道杂化作用对此也有一定的贡献。纵观全图,两吸附体系的态密度图总体类似,从而可以推断其吸附及成键方式的一致性,此结论与吸附构型分析的结果一致。

图4 CAL在M-Au(111)面上吸附的分波态密度(PDOS)和总态密度(TDOS)Fig.4 Partial densities of states(PDOS)and total densities of states(TDOS)of CALadsorption on M-Au(111)surfaces
差分电荷密度是一种能够直观描述吸附后体系电子重排的分析方法31。为了对CAL与M-Au (111)面的电子交互作用有更加鲜明的理解,本文计算了最稳定吸附构型的差分电荷密度,如图5所示。差分电荷密度定义为CAL吸附于M-Au(111)面前后电荷密度之差,即:Δρ=ρCAL/M-Au(111)-ρCAL-ρM-Au(111),其中ρCAL/M-Au(111)、ρCAL和ρM-Au(111)分别为体系的总电荷密度、CAL的电荷密度以及M-Au(111)面的电荷密度。图中蓝色区域代表失去电子,红色区域代表得到电子,且颜色越深、区域越大代表得失电子数越多。由图5可知,CAL在M-Au(111)面上的电子得失区域较为一致,从而进一步验证了CAL在M-Au(111)面上吸附方式的一致性。同时可以看出,CAL/Ir-Au(111)体系中的颜色区域及深度都略大于CAL/In-Au(111)体系,因此可知Ir-Au (111)面对于CAL的吸附力略大于In-Au(111)面,该结论与吸附能的分析结果相吻合。

图5 CAL在M-Au(111)面上的差分电荷密度Fig.5 Deformation density of CALadsorption on M-Au(111)surfaces
3.2.3 CAL的电子性质分析
为了能够更加深入了解CAL吸附前后的电子性质以及不同催化剂对其的影响,本文对吸附前以及吸附于M-Au(111)面上的CAL的分波态密度和Mulliken电荷布居进行了研究。
根据图4的分析可知,CAL的电子贡献主要来自于其p轨道,图6作出了CAL吸附前以及吸附于M-Au(111)面后的分波态密度图。从该图可知,当CAL吸附于M-Au(111)面后,CAL的分波态密度费明显降低,说明吸附后CAL向M-Au(111)面转移了大量电子。费米能级右侧的峰基本消失,说明吸附后CAL的反键π*轨道受到破坏。费米能级左侧的峰明显变小且向低能级移动,说明吸附后CAL的p轨道与金属表面发生强相互作用,形成σ键。相比较两类吸附体系,CAL在Ir-Au(111)面上的p轨道偏移和形变程度都略大于In-Au(111)面,进一步说明了CAL与Ir-Au(111)面的相互作用更强。其中,位于-6.53及-3.39 eV处的高峰主要来自于C=O的贡献4。

图6 CAL吸附前后p轨道的分波态密度Fig.6 p oribital partial density of states ofCALbefore and after adsorption
结合文献4报道以及表4可知,CAL的C=O相比较于C=C具有更强的极性,即C=O更有利于吸附于金属表面。对于In-Au(111)面和Ir-Au(111)面上吸附的CAL,其O1、C2、C3、C4以及分子整体所带电荷量分别为-0.34e、0.29e、-0.08e、0.02e、0.26e和-0.27e、0.29e、-0.08e、0.03e、0.36e。由此可以看出,吸附后CAL向金属表面转移电子,使得CAL带有一定量的正电荷,并且Ir-Au(111)面上CAL的转移电荷数相对更多,造成了CAL与该面更强的作用力。这可能是由于In和Ir原子都具有较大的有效核电荷,同时Ir原子的半径较In原子小,使得In-Au(111)面上的空轨道具有更强的电子对接受能力。相比较分子链上的电荷转移数可得,C=O的电荷转移数较C=C多,说明CAL的C=O与金属的作用更强。其中,C2原子上的电荷变化量相对细微,这可能是由于金属反馈部分电子到C=O上,并通过π键传递电子至C2原子上。
综上所述,在M-Au(111)面上,CAL最有可能通过C=O垂直吸附于M-Au(111)面的TopM位,且在Ir-Au(111)面上的吸附相对更稳定。同时对于稳定吸附构型的态密度、差分电荷密度以及Mulliken电荷布居等分析表明:当CAL以C=O稳定吸附于M-Au(111)面时,C=O能与金属表面发生强相互作用,并且CAL/Ir-Au(111)体系的电荷转移量较CAL/In-Au(111)体系更加显著,进一步说明了Ir-Au(111)面对于CAL的吸附作用强于In-Au(111)面。
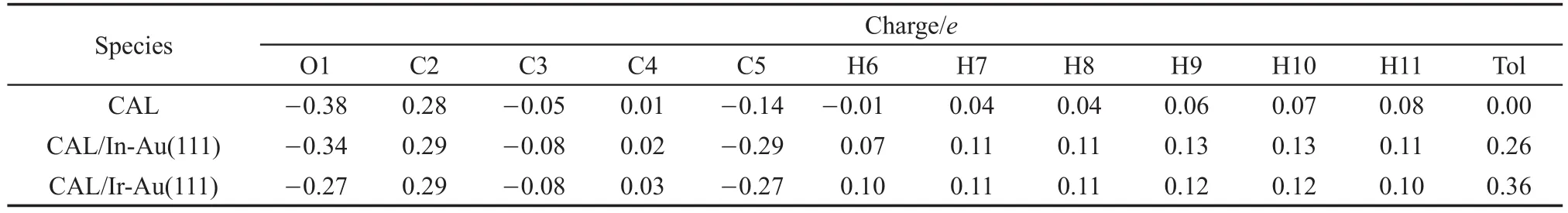
表4 CAL在M-Au(111)面上的Mulliken电荷布居数Table 4 Mulliken charges of CALadsorption on M-Au(111)surfaces
4 结论
本研究基于DFT,首先计算了M原子修饰Au (111)面形成的3种模型合金的稳定性,然后探讨了最稳定模型的化学活性以及CAL在其表面吸附的相关性质,结果如下:
(1)In原子修饰的In-Au(111)面的模型稳定性为(c)>(b)>(a)。对于以间位态存在的模型(c),其金属表面几何形变最小,形成能为8068.22 kJ·mol-1,结合能为391.71 kJ·mol-1,d带中心为-3.54 eV。Ir原子修饰的Ir-Au(111)面的模型稳定性为(a)>(b)>(c)。对于以聚集态存在的模型(a),其金属表面几何形变最小,形成能为10252.83 kJ·mol-1,结合能为425.78 kJ·mol-1,d带中心为-3.48 eV。
(2)CAL在In-Au(111)面和Ir-Au(111)面上的吸附,分别具有54种和70种初始吸附构型,经计算优化后能稳定存在的构型各自只有17种和22种,并且当CAL以C=O吸附于TopM位时,CAL形变最大,吸附构型最稳定,吸附能分别为145.45和152.21 kJ·mol-1,转移电荷量分别为0.26e和0.36e。其中,位于-7.04 eV至费米能级处的p、d轨道杂化,对于CAL在M-Au(111)面上的吸附具有重要贡献。
(3)分析比较In-Au(111)面和Ir-Au(111)面,发现后者不仅几何形变更小、形成能及结合能更大、构型更稳定以及d带中心更低,而且对于CAL的吸附,表现出更加优良的性能,即具有更大的吸附能、更低的态密度以及更明显的电子转移。因此,Ir-Au(111)面较In-Au(111)面的配体效应更佳。同时,相比较于Au(111)面,M原子的修饰改善了金属表面的性质及其对于CAL的吸附能力。
(1) Haruta,M.;Kobayashi,T.;Sano,H.Chem.Lett.1987,16,405. doi:10.1246/cl.1987.405
(2) Bus,E.;Prins,R.;Van Bokhoven,J.A.Catal.Commun.2007, 8,1397.doi:10.1016/j.catcom.2006.11.040
(3) Bailie,J.E.;Hutching,G.J.Chem.Commun.1999,21,2151. doi:10.1039/a906538e
(4) Jiang,J.H.;Xia,S.J.;Ni,Z.M.;Zhang,L.Y.Chem.J.Chin. Univ.2016,37,693.[蒋军辉,夏盛杰,倪哲明,张连阳.高等学校化学学报,2016,37,693.]doi:10.7503/cjcu20150844
(5) Aguirre,A.;Barrios,C.E.;Aguilar-Tapia,A.;Zanella,R.; Baltanás,M.A.;Collins,S.E.Top.Catal.2016,59,347. doi:10.1007/s11244-015-0425-6
(6) Ngoepe,P.N.M.;Meyer,W.E.;Auret,F.D.;Omotoso,E.; Diale,M.;Swart,H.C.;Duvenhage,M.M.;Coetsee,E.Physica B 2016,480,209.doi:10.1016/j.physb.2015.08.051
(7) Torres,C.;Campos,C.;Fierro,J.L.G.;Oportus,M.;Reyes,P. Catal.Lett.2013,143,763.doi:10.1007/s10562-013-1034-2
(8) Lyu,J.H.;Wang,J.G.;Lu,C.S.;Ma,L.;Zhang,Q.F.;He,X. B.;Li,X.N.J.Phys.Chem.C 2014,118,2594.doi:10.1021/ jp411442f
(9) Han,X.X.;Zhou,R.X.;Lai,G.H.;Zheng,X.M.Catal.Today 2004,93,433.doi:10.1016/j.cattod.2004.06.053
(10) Zhang,L.Y.;Shi,W.;Xia,S.J.;Ni,Z.M.Acta Phys.-Chim. Sin.2014,30,1847.[张连阳,施 炜,夏盛杰,倪哲明.物理化学学报,2014,30,1847.]doi:10.3866/PKU.WHXB201407141
(11) Cárdenas-Lizana,F.;Gómez-Quero,S.;Hugon,A.;Delannoy, L.;Louis,C.;Keane,M.A.J.Catal.2009,262,235. doi:10.1016/j.jcat.2008.12.019
(12) Huang,T.H.;Zhao,Y.J.;Tian,Z.F.;Li,X.L.;Liu,Q.;Zhao, D.Y.Acta Phys.-Chim.Sin.2014,30,2307.[黄天辉,赵玉娟,田兆福,李小兰,刘 茜,赵东元.物理化学学报,2014,30, 2307.]doi:10.3866/PKU.WHXB201410142
(13) Li,X.H.;Zheng,W.L.;Pan,H.Y.J.Catal.2013,300,9. doi:10.1016/j.jcat.2012.12.007
(14) Delbecq,F.;Li,Y.;Loffreda,D.J.Catal.2016,334,68. doi:10.1016/j.jcat.2015.10.028
(15) Yang,Q.Y.;Zhu,Y.;Tian,L.;Xie,S.H.;Pei,Y.;Li,H.;Li,H. X.;Qiao,M.H.;Fan,K.N.Appl.Catal.A-Gen.2009,369,67. doi:10.1016/j.apcata.2009.08.032
(16) He,X.;Chen,Z.X.;Kang,G.J.J.Phys.Chem.C 2009,113, 12325.doi:10.1021/jp9006729
(17) Rojas,H.A.;Martínez,J.J.;Díaz,G.;Gómez-Cortés,A.Appl. Catal.A-Gen.2015,503,196.doi:10.1016/j.apcata.2015.07.023 (18) Zhao,J.;Ni,J.;Xu,J.H.;Xu,J.T.;Cen,J.;Li,X.N.Catal. Commun.2014,54,72.doi:10.1016/j.catcom.2014.05.012
(19) Obot,I.B.;MacDonald,D.D.;Gasem,Z.M.Corrosion Sci. 2015,99,1.doi:10.1016/j.corsci.2015.01.037
(20) Yu,Y.X.ACS Appl.Mater.Interfaces 2014,6,16267. doi:10.1021/am504452a
(21) Manyer,H.G.;Yang,B.;Daly,H.;Moor,H.;McMonagle,S.; Tao,Y.;Yadav,G.D.;Goguet,A.;Hu,P.;Hardacre,C. ChemCatChem 2013,5,506.doi:10.1002/cctc.201200447
(22) Mohr,C.;Hofmeister,H.;Radnik,J.;Claus,P.J.Am.Chem. Soc.2003,125,1905.doi:10.1021/ja027321q
(23) Tao,J.;Yao,Z.J.;Xue,F.Fundamentals of Material Science;Chemical Industry Press:Beijing,2006;pp 50-51.[陶 杰,姚正军,薛 烽.材料科学基础.北京:化学工业出版社,2006: 50-51.]
(24) Chang,C.R.;Long,B.;Yang,X.F.;Li,J.J.Phys.Chem.C 2015,119,16072.doi:10.1021/acs.jpcc.5b03965
(25) Amaya-Roncancio,S.;Linares,D.H.;Sapag,K.;Rojas,M.I. Appl.Surf.Sci.2015,346,438.doi:10.1016/j. apsusc.2015.03.215
(26) Pirillo,S.;López-Corral,I.;Germán,E.;Juan,A.Vacuum 2014, 99,259.doi:10.1016/j.vacuum.2013.06.013
(27) Yang,Y.J.;Teng,B.T.;Liu,Y.;Wen,X.D.Appl.Surf.Sci. 2015,357,369.doi:10.1016/j.apsusc.2015.09.039
(28) Haubrich,J.;Loffreda,D.;Delbecq,F.;Jugnet,Y.;Sautet,P.; Krupski,A.;Becker,C.;Wandelt,K.Chem.Phys.Lett.2006, 433,188.doi:10.1016/j.cplett.2006.10.123
(29) Delbecq,F.;Sautet,P.J.Catal.2003,220,115.doi:10.1016/ S0021-9517(03)00249-5
(30) Zeinalipour-Yazdi,C.D.;Willock,D.J.;Machado,A.;Wilson, K.;Lee,A.F.Phys.Chem.Chem.Phys.2014,16,11236. doi:10.1039/c3cp53691b
(31) Shi,W.;Zhang,L.Y.;Xia,S.J.;Ni,Z.M.Acta Phys.-Chim. Sin.2014,30,2249.[施 炜,张连阳,夏盛杰,倪哲明.物理化学学报,2014,30,2249.]doi:10.3866/PKU.WHXB201408283
Comparison of Properties of In-Au(111)and Ir-Au(111)Alloy Surfaces, and Their Adsorption to Crotonaldehyde
JIANG Jun-Hui QIAN Meng-Dan XUE Ji-Long XIASheng-Jie NI Zhe-Ming*SHAO Meng-Meng
(College of Chemical Engineering,Zhejiang University of Technology,Hangzhou 310014,P.R.China)
The stability of M-Au(111)surfaces modified by M(M=In,Ir)was investigated using density functional theory.The most favorable model was selected to explore the chemical reactivity and adsorption of crotonaldehyde.The stability of the M-Au(111)surfaces was calculated using geometric configuration,and formation and cohesive energies.The calculations showed that the stability of the In-Au(111)surface increased as the atomic spacing of In was increased.Conversely,the Ir-Au(111)showed the opposite trend.The adsorption at the TopMsite was most stable when the crotonaldehyde on the M-Au(111)surfaces interacting via the C=O. Additionally,the adsorption energies were at their maximum.A combination of structural changes,density of state,deformation density and Mulliken charge analysis showed that the deformation of crotonaldehyde was larger than other adsorption modes,with an obvious charge transfer.Additionally,p and d orbital hybridization between-7.04 eV to Fermi level was found to have an important contribution to the adsorption process. Compared with the Au(111)surface,the stability and adsorption capacity of the M-Au(111)surfaces were significantly improved following modification by M atoms.Importantly,the Ir-Au(111)surface was found to have higher stability and activity,and stronger adsorption of crotonaldehyde than the In-Au(111)surface.
In-Au(111)surface;Ir-Au(111)surface;Modification;Crotonaldehyde;Adsorption; Density functional theory
O641
10.3866/PKU.WHXB201609302
Received:July 13,2016;Revised:September 30,2016;Published online:September 30,2016.
*Corresponding author.Email:jchx@zjut.edu.cn;Tel:+86-13858123256.
The project was supported by the National Natural Science Foundation of China(21503188).
国家自然科学基金(21503188)资助项目
猜你喜欢
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
婚姻与家庭·性情读本(2017年3期)2017-04-27 23:13:07
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
航天返回与遥感(2014年4期)2014-07-31 17:47:47
健康必读(2014年2期)2014-06-23 09:12:26
无机化学学报(2014年4期)2014-02-28 17:31:08
