
利用SRAP和SSR标记构建红麻遗传连锁图谱
2016-12-24 06:55武耀龙李德芳李建军黄思齐李辉唐慧娟陈安国
中国麻业科学 2016年6期
武耀龙,李德芳,李建军,黄思齐,李辉,唐慧娟,陈安国
(中国农业科学院麻类研究所,长沙410205)
利用SRAP和SSR标记构建红麻遗传连锁图谱
武耀龙,李德芳,李建军,黄思齐,李辉,唐慧娟,陈安国*
(中国农业科学院麻类研究所,长沙410205)
构建红麻遗传连锁图谱,为今后红麻重要农艺性状QTL定位、QTL图位克隆、优良基因筛选、分子标记辅助育种奠定良好的研究基础。对256对SRAP引物和64对棉花SSR引物进行了两次引物筛选,以泰红763×F71为作图群体亲本,150个F2代单株作为作图群体,构建红麻遗传连锁图谱。共筛选出条带清晰、多态性高的SRAP引物73对、SSR引物8对,构建的红麻遗传连锁图谱全长2155.43 cM,平均间距为16.09 cM,含有128个多态性标记,分布于18个连锁群。本研究初步证实了棉花SSR引物用于红麻是可行的,构建的红麻遗传连锁图谱密度较高,标记较为均匀,适合后续的QTL定位、QTL图位克隆等研究工作。
遗传连锁图谱;红麻;SRAP;SSR
红麻(Hibiscus cannabinus L.),又名洋麻、槿麻、钟麻等,属于锦葵(Malvaceae)木槿属(Hibiscus)一年生韧皮纤维作物,栽培种红麻为二倍体(2n=36)[1]。红麻的韧皮纤维柔软,纤维拉力强,其自然纤维具有抑菌、透气、吸湿性好、散失水分快、可降解等特性,用来生产汽车内衬、麻塑、纸地膜、轻型板材、绒毛浆、活性炭及环境友好型吸附材料,是重要的麻纺工业原料作物。因而红麻被视为二十一世纪有潜力的优势作物[2,3]。遗传连锁图谱(genetic linkage map)是研究优良性状基因定位的重要工具,同时也是研究基因组进化的基础[4]。较高密度的遗传图谱,在数量性状基因定位、图位克隆、重要农艺性状基因、种质资源鉴定和分子标记辅助选择育种等研究中发挥了十分重要的作用[5]。因此构建红麻遗传连锁图谱将推动红麻分子标记辅助育种研究的开展,同时也为高产、优质基因的筛选提供遗传基础。
Botstein等1980年首次研究了利用重组DNA探针得到的DNA多态性标记位点构建人类遗传连锁图谱之后,遗传连锁图谱逐步成为了研究生物基因组的重要工具,并且这一研究工具逐步开始在农作物中应用,先后有水稻[6]、大豆[7]、大麦[8]等作物的高密度连锁图谱的构建成功。与这些农作物相比,麻类作物在遗传连锁图谱的构建这方面研究起步较晚,分子遗传的基础研究比较滞后,Liu等[9]利用SSR引物构建了一张含有132个标记,全长为2265.1 cM的第一张苎麻遗传连锁图谱;福建农林大学在红麻分子遗传连锁图谱构建研究方面起步较早,张广庆等[10]、张晓琛等[11]应用ISSR、SRAP、RAPD分子标记以Ga42和阿联红麻为亲本分别构建了一张含有78和134个标记的红麻遗传连锁图谱,陈美霞等[12]以阿联红麻和福红992为亲本,利用SRAP、ISSR、RAPD构建了一张含有26个连锁群共307个标记、总长为4924.8 cM的红麻连锁图谱。
本研究在主要采用SRAP分子标记的基础之上,同时采用了棉花的SSR引物用于红麻遗传连锁图谱的构建,对棉花SSR引物用于红麻分子标记的可行性进行了初步探讨,同时不同类型分子标记技术用于构建连锁图谱将有助于高质量红麻图谱的构建。本研究使用SRAP引物和棉花SSR引物构建红麻遗传连锁图谱将为日后进一步加密图谱、红麻产量及品质等相关性状的QTL定位、红麻优良基因的筛选、QTL图位克隆、红麻分子标记辅助育种的开展奠定良好的研究基础。
1 材料与方法
1.1 作图群体的构建
为保证实验的进行性以及实验结果的可靠性,本实验选取了6对亲本杂交组合,分别为:泰红763×F71、泰红763×83-20、泰红763×台农1号、Y1A1(中红麻13号)×F71、Y1A1×83-20和泰红763×83-18。本实验所用的6个红麻亲本来自于中国农业科学院麻类研究所黄红麻种植资源课题组(表1)
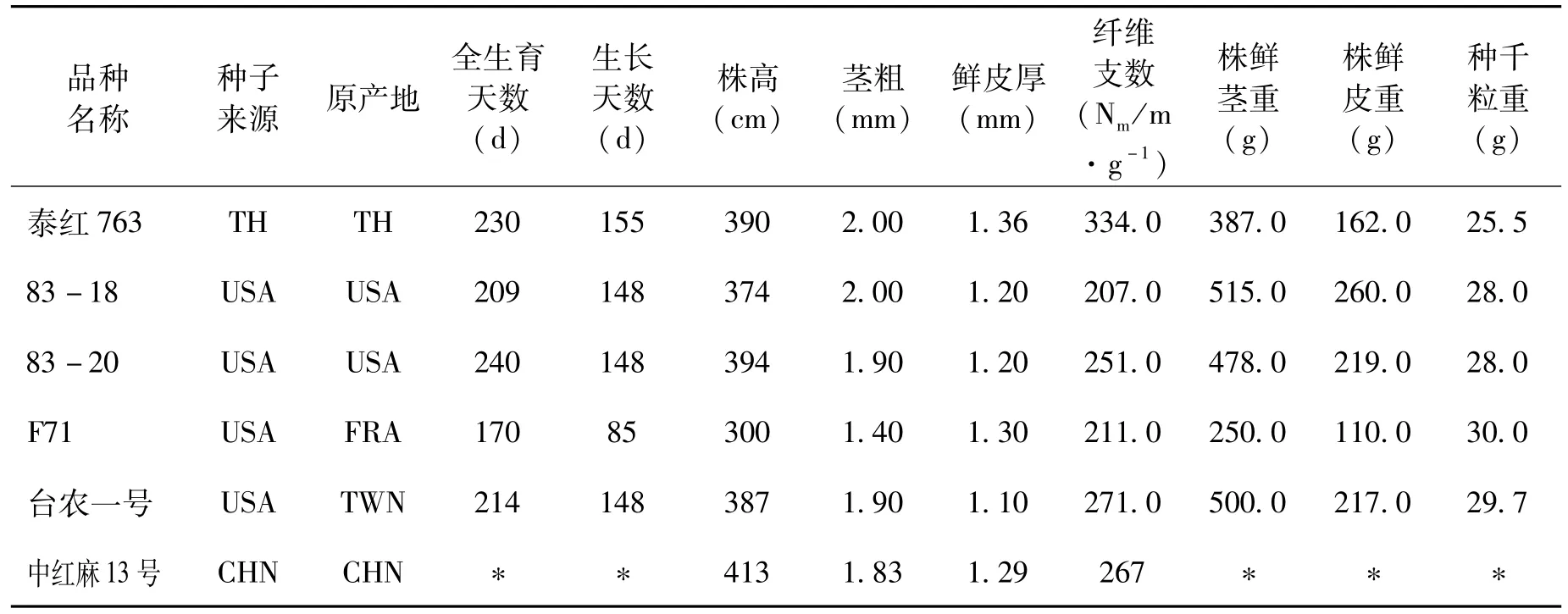
表1 供试红麻材料Tab.1 Experimental kenaf materials
1.2 基因组DNA提取
亲本和F1代的DNA均提取自红麻子叶,F2代单株DNA提取自红麻苗期的幼嫩叶片。采用北京天根公司生产的植物基因组DNA提取试剂盒(离心柱型)提取DNA,并用1%琼脂糖凝胶电泳检测DNA质量,Unico公司生产的UV-2100型紫外分光光度计检测DNA浓度,稀释到20 ng/μL,置于-20℃冰箱保存备用。
1.3 SRAP和SSR分子标记分析
256对SRAP引物来自于Li和Quiros[13]、Lin[14]等及Riaz[15]等发表的SRAP引物序列(表2)。64对SSR引物(表3)选自Cotton DB数据库(http://algodon.tamu.edu/~mapbase/SSR-framepage.htm)。所有的引物均由上海生工生物股份有限公司合成。
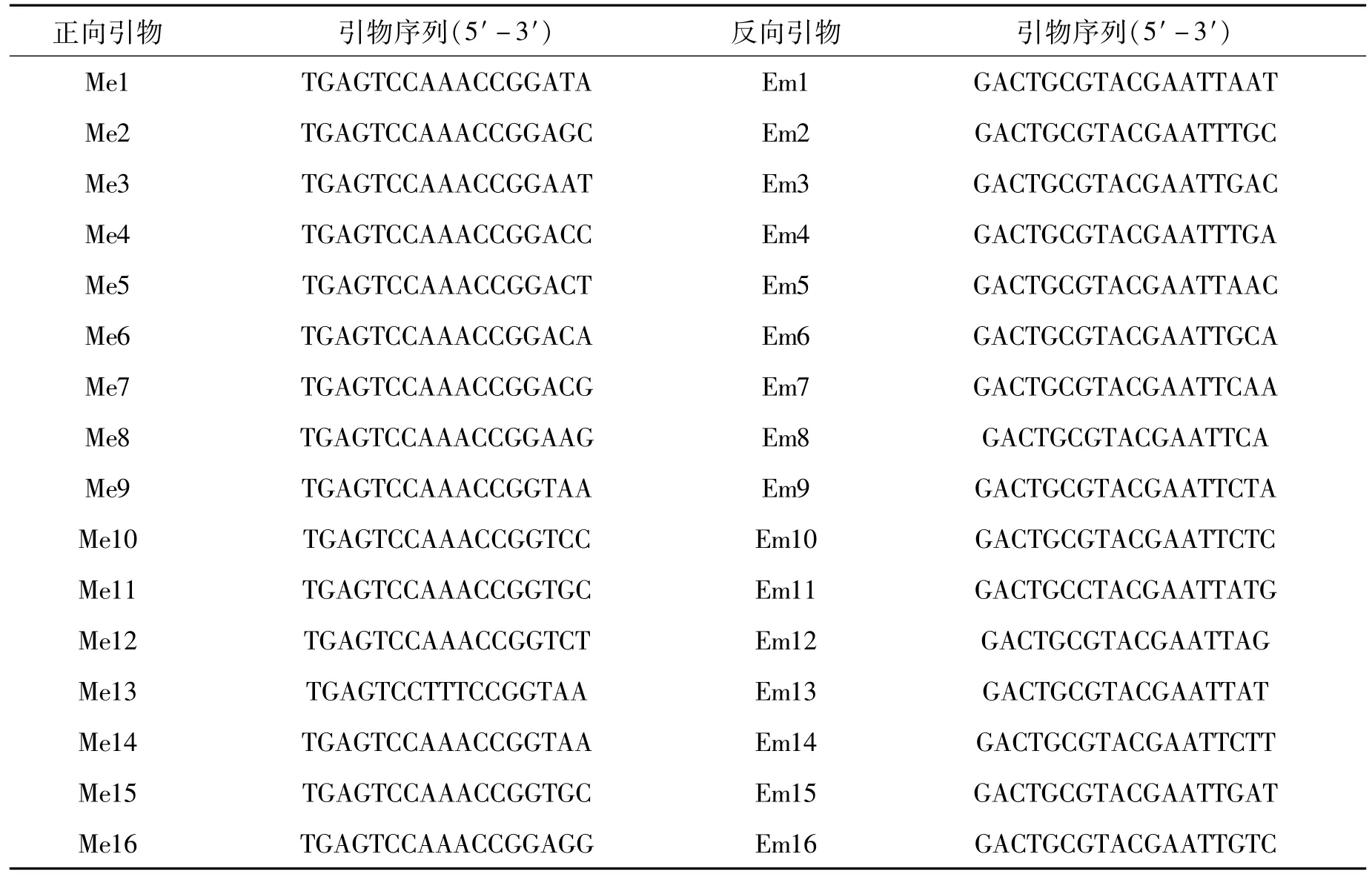
表2 SRAP引物序列Tab.2 Primer sequences of SRAP
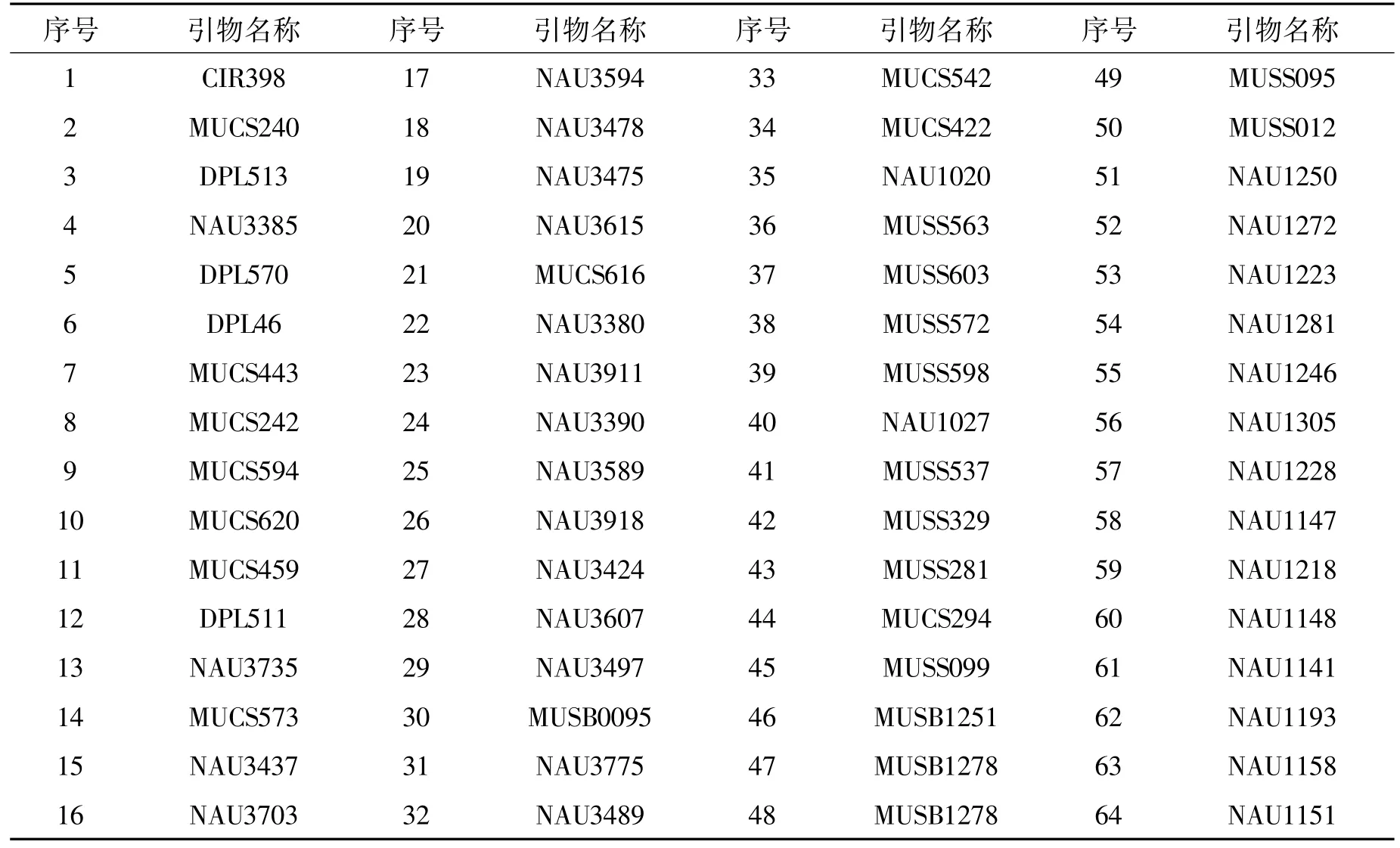
表3 SSR引物列表Tab.3 List of SSR primer
SRAP反应程序:94℃预变性5 min;94℃变性1 min,37℃退火1 min,72℃延伸1 min,5个循环;94℃变性1 min,55℃退火1 min,72℃延伸1 min,32个循环;循环结束后,72℃延伸10 min,产物于4℃保存。
SSR反应程序:94℃变性4 min;94℃变性30 s,52℃复性30 s,72℃延伸1 min,35个循环;72℃延伸10 min;4℃保存。SSR反应程序参考陈浩东[16]、王志伟[17]等的反应程序的基础上作了适当的调整。
扩增产物在8%聚丙烯酰胺凝胶中分离,电压200 V、电流220 mA的条件下电泳1 h,快速银染检测。本实验所使用PCR仪为BIO-RAD S1000型,电泳仪为北京君意JY-JX7型,电泳仪电源为BIO-RAD PowerPac Basic。
SRAP-PCR与SSR-PCR反应体系参考之前的研究[18]:2 μL 10×Taq Reaction Buffer、20 ng模板DNA、dNTP 220 μmol/L、引物0.35 μmol/L、Taq DNA聚合酶0.5U,总体积为20 μL。
1.4 标记命名与数据记载
读取条带是在相同迁移位点上的多态性条带记为“1”,无条带的记为“0”。为保证实验的重复性与可靠性,条带模糊不清的舍弃该条带,胶片出现大面积条带模糊或条带跑偏等现象的舍弃该胶片进行重复性实验。
本研究中出现的标记名称前面一部分为引物名称或引物名称简写,后面的数字表示该标记DNA片段的大小,中间用“-”连接。例如,M1E3-561表示SRAP引物Me1Em3扩增出来的大小为561 bp的条带;MUCS242-687表示棉花SSR引物MUCS242扩增出来的687 bp的多态性条带。条带片段的大小的计算本实验采用对天根公司生产的100 bp marker每一条条带到点样孔之间的距离和条带片段大小作拟合曲线(图1),对电泳出来的每一板胶,统计其100 bp片段与点样孔之间的间距,从而对拟合曲线进行参数矫正,然后统计每一个多态性条带与点样孔之间的间距,利用Excel计算出具体片段大小。该方法的基本原理是DNA片段在PAGE凝胶中在电场下的移动主要是依靠电荷效应和分子筛效应,因此电泳的迁移率与分子量的对数值呈线性关系,相对迁移率=样品迁移距离/标记迁移距离,据此可以计算出未知DNA片段大小。
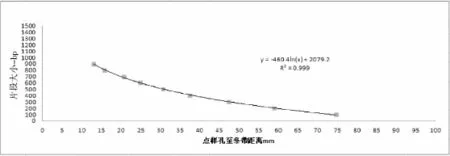
图1 根据marker片段大小所做曲线计算DNA多态性片段大小Fig.1 Calculation of DNA polymorphism fragment with curve made by marker MW
1.5 软件参数设定
按照JoinMap 4.0[19]软件的要求,所有标记母本泰红763的带型记为a,父本F71的带型记为b;如果该条带泰红763对F71为显性,则F2代中显性的记为a,隐性的记为c;如果该条带F71对泰红763为显性,则F2代中显性的计为b,隐性的计为d;杂种带型记为h。由于某些原因造成条带模糊不清或数据缺失的记为“-”。
参考盛洁[20]等的方法利用Excel软件对分子标记的分离数据进行偏分离卡方检验,计算标记是否符合3:1的分离比例,若卡方值X2(0.05)>3.84则认为差异显著。
JoinMap 4.0软件构建遗传连锁图谱,软件的Lod值设为3.0,重组频率为0.25,采用Kosambi函数计算图距。JoinMap软件构建图谱过程中,将间距大于100 cM的连锁群进行拆分处理,间距过大的单个标记舍弃。连锁群的命名依据连锁群的长度大小顺序分别用LG(linkage group)后面加数字序号命名连锁群,如:LG1表示第一连锁群。
2 结果与分析
2.1 作图亲本的确立
本研究作图杂交亲本组合为泰红763×F71,选择依据为后续SRAP-PCR对6个亲本组合的扩增结果(表4)与两亲本表型性状的差异。结果表明,组合泰红763×F71不仅表型性状的差异巨大,对两亲本SRAP分子标记扩增的结果也表明二者之间差异较大,适合作为连锁图谱构建亲本。
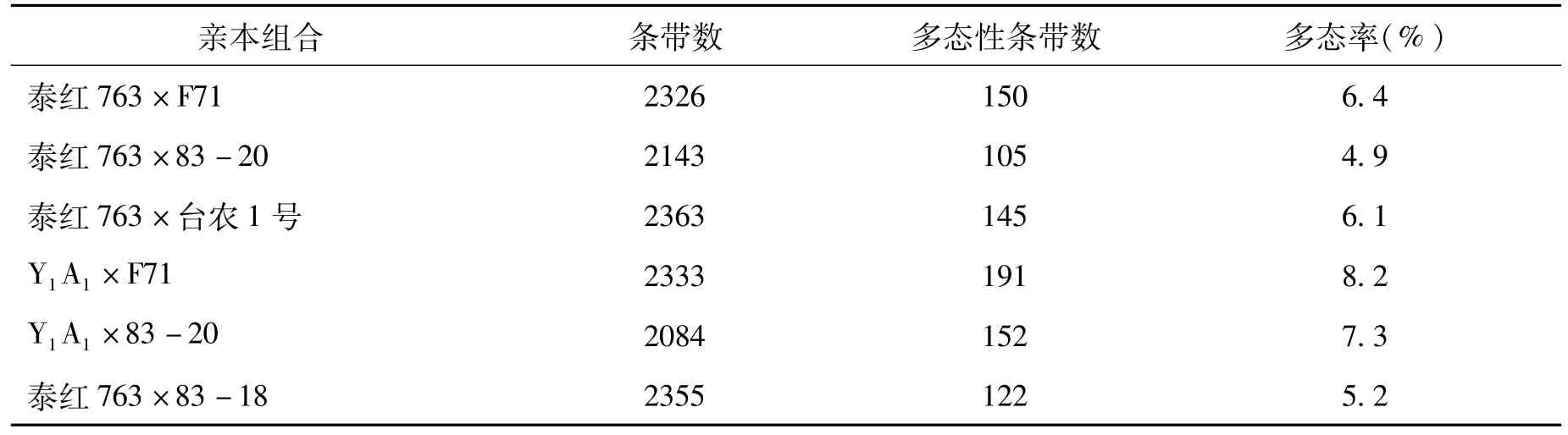
表4 6个亲本扩增结果Tab.4 SRAP results of six materials
2.2 SRAP和SSR结果分析
第一次引物筛选以6个亲本材料DNA为模板进行SRAP-PCR扩增,引物的再次筛选以亲本泰红763、F71以及10个随机选取的F2代群体单株DNA为模板对引物进行SRAP-PCR、SSRPCR扩增;由于本实验使用的SSR引物较少,因此直接进行了第二次引物筛选,跳过了引物的初次筛选。
初次引物筛选结果(表5):在256对引物组合中,条带主要分布于200~800 bp之间,共有175对引物能够扩增出多态性条带,占总数的68.3%;256对引物共扩增出2573条条带,平均每个引物可扩增出10.1条;175对引物共扩增出多态性条带467个,平均每个引物可扩增出2.67条多态性条带,多态率为18.2%;从256对引物中共筛选出91对带型清晰明亮且多态性较高的引物。
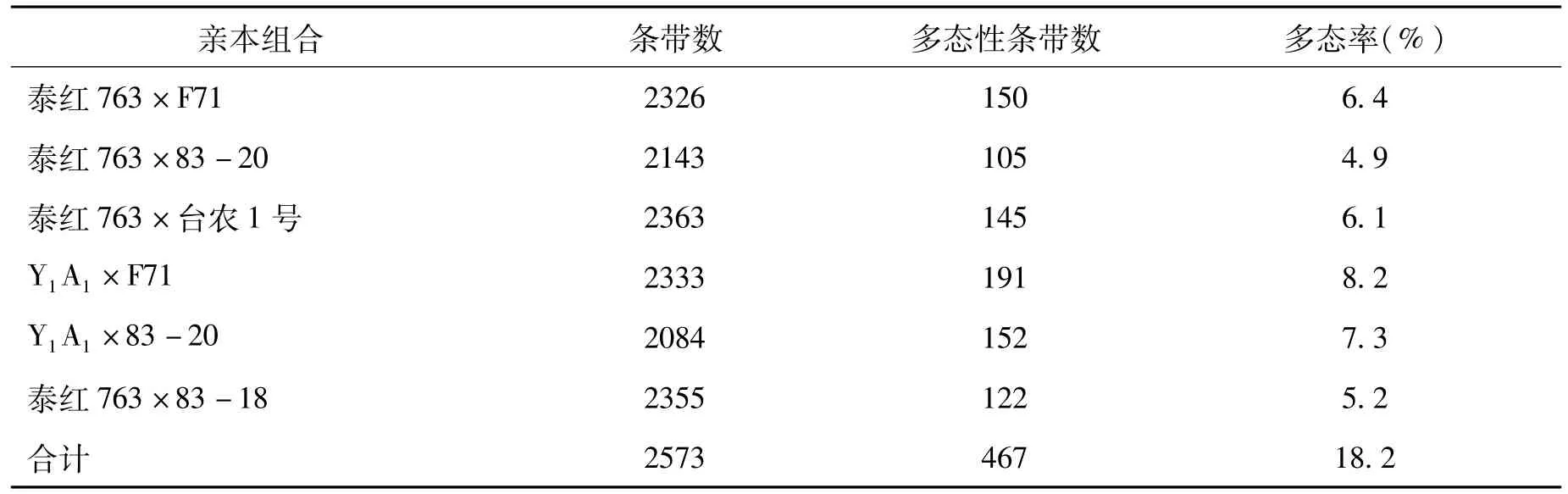
表5 初次引物筛选数据统计Tab.5 Statistics of first round primer screening
二次引物筛选结果(表6):初次筛选出来的91对多态性引物中有11对引物没有扩增出多态性条带,占总数的12%;所有引物共扩增出1487条条带,平均每个引物可扩增16.3条条带。多态性条带共有274条,80对能够扩增出条带的引物平均每对扩增3.4条多态性条带,多态率为18.4%。64对SSR引物有57对引物能够扩增出条带,其中有36对引物有多态性条带,条带主要分布在300~700 bp之间,共扩增出864条条带,其中89条为多态性条带,多态率为10.24%。引物的再次筛选共筛选出带型清晰、易辨认且多态性较好的SRAP引物73对、SSR引物8对。
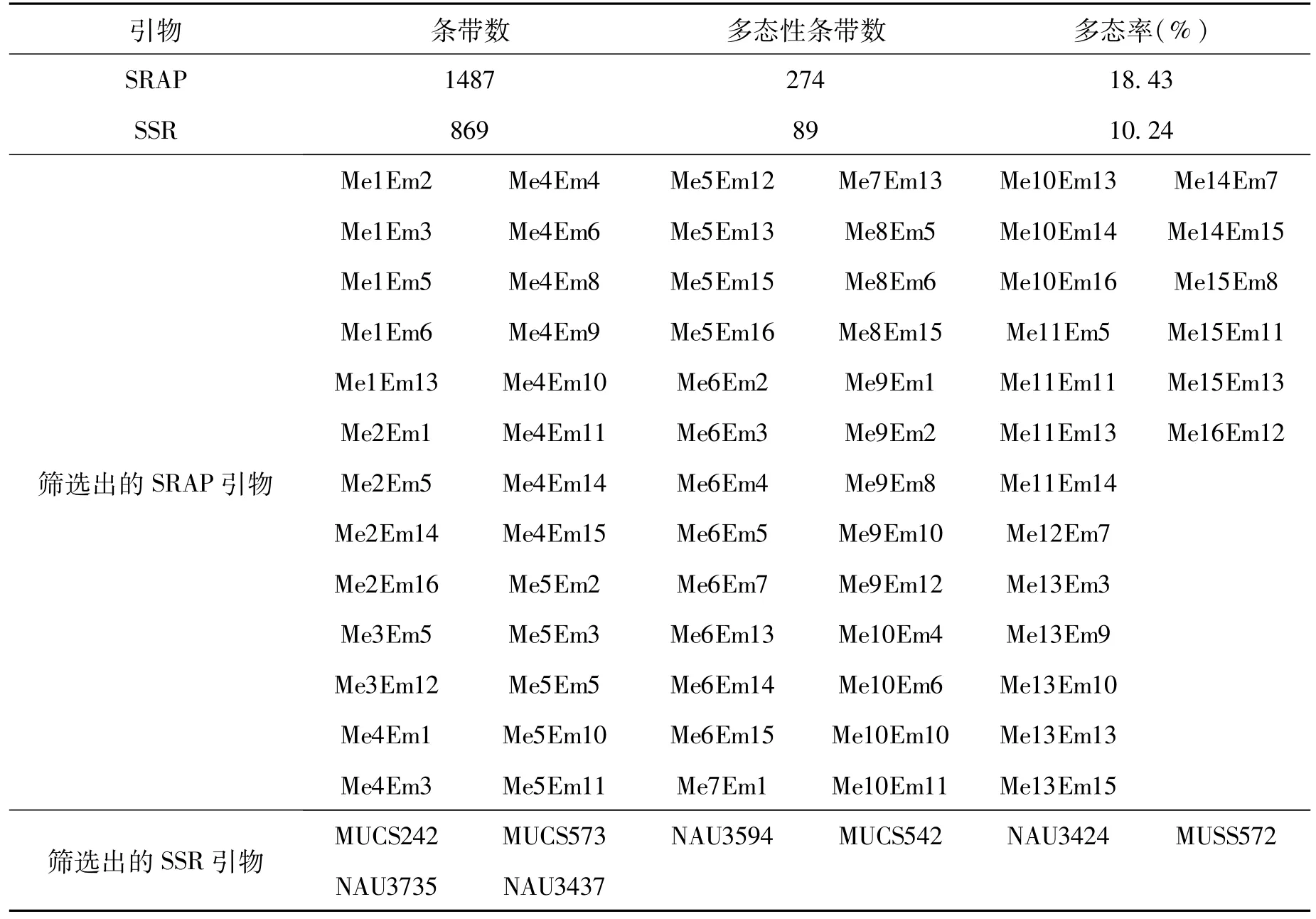
表6 二次引物筛选结果Tab.6 Results of second round primer screening
本实验对共231个多态性标记为点进行遗传连锁分析,最后共有128个标记进入18个连锁群,其中有120个SRAP标记和8个SSR标记,有103个多态性标记没有进入连锁图谱。至少含有5个标记的连锁群有10个,8个连锁群只有2~3个多态性标记,说明该图谱还需要更多的标记添加进去,有待后续实验的完善。整张图谱覆盖基因组的长度为2155.43 cM,两个标记间的平均距离为16.09 cM,最大间距为78.223 cM,最小间距为0;最长的LG1长达840.424 cM,含有40个标记,最短的LG18只有6.116 cM;图谱标记间距较为均匀,有一小部分区域出现标记聚集现象。
128个多态性标记以及偏分离标记在连锁图谱的分步情况如表7和图2:
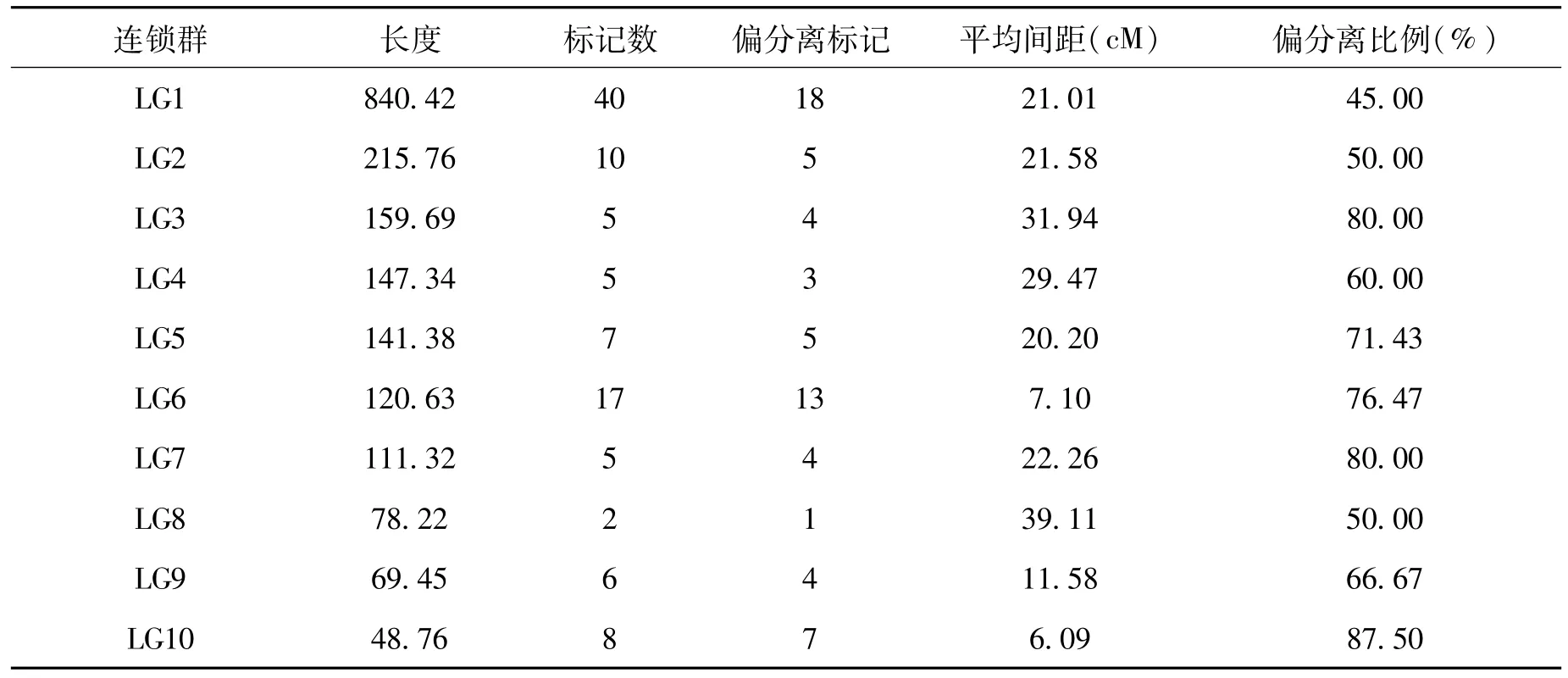
表7 128个标记在连锁图谱上的分布Tab.7 Distribution of 128 loci on the genetic map
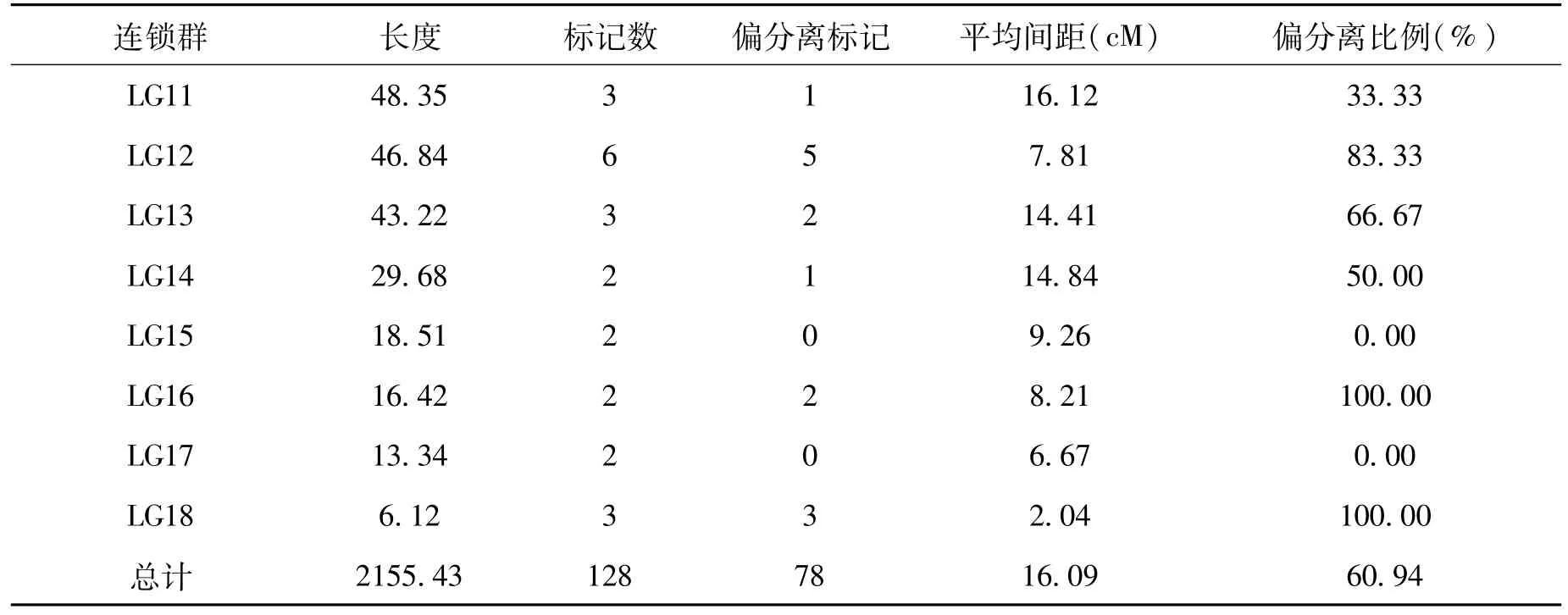
连锁群长度标记数偏分离标记平均间距(cM)偏分离比例(%)LG1148.353116.1233.33 LG1246.84657.8183.33 LG1343.223214.4166.67 LG1429.682114.8450.00 LG1518.51209.260.00 LG1616.42228.21100.00 LG1713.34206.670.00 LG186.12332.04100.00总计2155.431287816.0960.94

图2 利用128个多态性标记构建的红麻遗传连锁图谱Fig.2 kenaf genetic linkage map made by 128 polymorphic markers
3 讨论
3.1 SSR引物通用性
SSR(Simple Sequence Repeat)是一种基于PCR反应的分子标记技术,具有操作简便、高多态性、高共显性等特点,广泛地应用于动物、植物、微生物等研究领域[21]。但其缺点是扩增位点较少,引物的开发成本高且SSR标记中所使用的引物不同,实验结果差异较大。分子标记技术种间的通用性有利于提高引物利用效率,降低开发成本[22]。近年来,众多研究发现SSR引物在近缘物种乃至远源物种中的通用性得到了较好的验证,Sharma等[23]分析发现98对水稻SSR引物和20对甘蔗EST-SSR引物在23份竹子中的通用性分别为44.9%和75%;钟敏等研究发现1205对绿豆基因组DNA SSR引物在小豆、豇豆和饭豆中的通用性比例分别为50%、73.3%和81.6%,多态性比例分别为4.1%、1.7%和1.5%。郑丽珊等[24]利用1595对棉花SSR引物对不同基因型的贡蕉和野蕉进行扩增,得到183对有效扩增引物。红麻和棉花一样都属于锦葵科木槿属作物,因此本研究选取了64对棉花SSR引物对红麻作图群体双亲和10个F2代单株进行PCR扩增,其中57对引物扩增出了条带,通用性比例高达89%;36对引物具有多态性条带,多态性比例为56%;共选出8对多态性较高的引物用于作图群体的扩增,很好地证明了棉花SSR引物用于红麻分子标记研究是可行的,同时也初步证实了近缘物种间进行SSR引物的通用性分析是有效可行的。
3.2 多种分子标记技术对构建连锁图谱的影响
多种分子标记技术的共同使用构建物种的遗传连锁图谱成为了近年来遗传图谱构建的趋势,王志伟等[17]利用AFLP与SSR分子标记技术构建了一张含有251个标记的棉花遗传连锁图谱,骆晚侠等[25]利用SSR和EST-SSR构建了一张含有145个标记的小豆遗传连锁图谱,Xia等[26]使用SNPs与SSR标记技术构建了第一张含269个标记的草鱼遗传连锁图谱。SRAP分子标记对开放阅读框的扩增效果较好,但对染色体末端的扩增较差;SSR标记的多态性高,重复性好,但同时扩增片段少,基因型判断错误概率大;RAPD分子标记不依赖于种属特异性和基因组的结构,一套引物可以分析不同生物基因组,但其重复性差,为显性标记,无法识别杂合子[27]。多种分子标记技术的使用有利于克服各种分子标记技术的缺陷,构建出来的遗传连锁图谱精细程度高、覆盖染色体上的基因范围更广。本实验中发现,SSR引物绝大多数都处于连锁群的中下部,如NAU3437的三个标记处于LG1的中下部;MUCS573-763处于LG2的中下部;NAU3424的两个标记处于LG6的下部;MUCS573-751处于LG5的中部,只有NAU3424-377处于所在连锁群的上部。陈美霞等[12]用SRAP、ISSR、RAPD构建的红麻遗传连锁图谱发现图谱中LG20、LG21、LG22、LG25、LG26仅仅包含有RAPD标记。造成一种标记集中分布在一个连锁群的同一位置或者同一连锁群上的原因可能是分子标记位点较少,标记的间隔过大引起的,也有可能是分子标记技术本身的特性决定的,因此在构建图谱过程中,应尽量多地开发高质量的引物并且多种分子标记技术共同使用,对提高图谱的精度,构建红麻高密度的饱和连锁图谱有重要的作用。
3.3 偏分离标记对连锁图谱构建的影响
植物中的偏分离标记是普遍存在的,普遍认为偏分离是物种进化强有力的推进力,对生物体有性繁殖的诸多要素是非常重要的,这些因素包括性别和性别比、重组、异型性染色体、生殖隔离和配偶选择等[28]。产生偏分离的原因可能与雌配子体、雄配子体、合子选择、作物种间群体配子重组率差异、偏分离研究与高密度分子标记遗传连锁图谱构建着丝点区域、遗传搭车效应、亲本遗传背景、群体类型和非遗传因素等有关[29]。在连锁图谱中,偏分离标记对图谱的准确性产生影响,不仅可以影响标记之间的距离,甚至可以影响到标记在连锁图谱上的顺序[30]。本实验中的128个标记中有78个(占60.9%)标记是偏分离标记,偏分离比例较为严重,几乎每个连锁群上都具有偏分离标记,只有LG15与LG17连锁群上没有偏分离标记,偏分离标记多以成簇的形式分布于连锁群的中部和两端,而且这些偏分离标记在一个聚集簇中大多按照一个方向偏离,或向母本偏离,或向父本偏离,余渝等[29]在棉花高密度遗传连锁图谱的构建中也同样发现了这种偏分离标记聚集的现象。在偏分离标记中有46个(58.9%)偏向父本基因型,32个(41.1%)标记偏向母本基因型。张广庆等[10],陈美霞等[12],张晓琛等[11]利用同样的作图亲本构建的三张连锁图谱偏分离比例都较高,分别为56%、26%和46%。而且在这三个图谱中标记数越多,偏分离比例就越小,标记数最多的为307,但偏分离标记数仍高达80个。因此可能是由于作图亲本的剩余杂合性导致等位基因型复杂,连锁关系难以确定是本次实验中图谱标记中偏分离比例较高的最主要原因[31]。当然偏分离标记比例较高同样与标记基因型的统计,亲本基因的突变等多方面因素有关。克服偏分离标记的方法最好是作图时先将偏分离的标记剔除,然后再将偏分离标记添加进去,观察偏分离标记对图谱的影响,最终确定标记的去留。但对偏分离标记的定位和遗传效应研究都必须建立在高密度遗传图谱的基础之上,本实验由于红麻分子遗传研究基础较为薄弱,时间有限,无法筛选更多的引物更多的标记进入连锁图谱,因此没有对偏分离标记做比对处理。随着红麻分子标记研究的不断进展,会有高密度的遗传图谱构建出来,对偏分离标记的定位也将成为可能。对偏分离标记的研究不仅有利于获得高精度的遗传图谱和QTL定位分析,也有利于红麻的分子育种的研究进展。
4 结论
本研究采用SRAP引物以棉花SSR引物构建出一张密度较高、分布较为均匀的红麻遗传连锁图谱,图谱含有128个多态性标记,分布于18个连锁群,全长为2155.43 cM,标记间平均间距为16.09 cM。
[1]谢晓美.红麻种质资源亲缘关系的形态学、细胞学与分子标记研究[D].中国农业科学院,2007.
[2]熊和平主编.麻类作物育种学[M].北京:中国农业科学技术出版社,2008.
[3]陶爱芬,张晓琛,祁建民.红麻综合利用研究进展与产业化前景[J].中国麻业科学,2007,01:1-5.
[4]Botstein D,White R L,Skolnick M,Davis R W.Construction of a genetic linkage map in man using restriction fragment length polymorphisms[J].American Journal of Human Genetics,1980,32.
[5]陈晖,陈美霞,陶爱芬,张广庆,徐建堂,祁建民,方平平.长果种黄麻SRAP标记遗传连锁图谱的构建及3个质量性状基因定位[J].中国农业科学,2011,12:2422-2430.
[6]Harushima Y,Yano M,Shomura A,et al.A high-density rice genetic linkage map with 2275 markers using a single F2population[J].Genetics,1998,148(1):479-494.
[7]Cregan P B,Jarvik T,Bush A L,et al.An integrated genetic linkage map of the soybean genome[J].Crop Science,1999,39(5):1464-1490.
[8]Ramsay L,Macaulay M,Degli Ivanissevich S,et al.A simple sequence repeat-based linkage map of barley[J].Genetics,2000,156(4):1997-2005.
[9]Liu T,Tang S,Zhu S,et al.QTL mapping for fiber yield-related traits by constructing the first genetic linkage map in ramie(Boehmeria nivea L.Gaud)[J].Molecular Breeding,2014:1-10.
[10]张广庆.应用SRAP和ISSR分子标记构建红麻的遗传连锁图谱[D].福建农林大学,2007.
[11]张晓琛.红麻分子遗传连锁图谱的构建[D].福建农林大学,2008.
[12]陈美霞.红麻遗传连锁图谱构建及重要性状基因定位与SCAR标记的开发[D].福建农林大学,2011.
[13]Li G,Quiros CF.Sequence-related amplified polymorpgism(SRAP)a new maker system based on a simple PCR reaction:its application to mapping and gene tagging in Brassica[J].Theor Appl Genet,2001,103:455-461.
[14]Lin Z,He D,Zhang X.Linkage map construction and mapping QTL for cotton fiber quality using SRAP,SSR and RAPD[J].Plant Breeding,2005,124:180-187.
[15]Riaz A,Li G,Quresh Z,et al.Genetic diversity of oilseed Brissica napus inbred based on sequence-related amplified polymorphism and its relation to hybrid performance[J].Plant Breeding,2001,120:411-415.
[16]陈浩东,李育强,洪亚辉.棉花SSR-PCR反应体系的优化[J].分子植物育种,2007,S1:182-186.
[17]王志伟.棉花分子遗传图谱构建和纤维品质QTL定位[D].河北农业大学,2007.
[18]武耀龙,李德芳,黄思齐,李建军,唐慧娟,陈艳翠,陈安国.红麻SRAP-PCR反应体系优化及多态性引物的筛选[J].中国麻业科学,2014,01:1-7.
[19]Van Ooijen J W.JoinMap 4[J].Software for the calculation of genetic linkage maps in experimental populations.Kyazma BV,Wageningen,Netherlands,2006.
[20]盛洁,胡建华.Excel软件的统计功能在卡方检验中的应用[J].医学信息,2008,01:28-31.
[21]赵海燕,渠云芳,黄晋玲.SSR分子标记在棉花遗传育种中的应用及进展[J].中国农学通报,2009,22:57-61.
[22]钟敏,程须珍,王丽侠,王素华,王小宝.绿豆基因组SSR引物在豇豆属作物中的通用性[J].作物学报,2012,02:223-230.
[23]Sharma R K,Gupta P,Sharma V,et al.Evaluation of rice and sugarcane SSR markers for phyloge-netic and genetic diversity analyses in bamboo.Genome.2008.
[24]郑丽珊,袁有禄,王静毅,冀小蕊,黄秉智,武耀廷.棉花SSR分子标记在香蕉中通用性的研究[J].分子植物育种,2007,05:667-672.
[25]骆晚侠,张李,杨凯,李奕松,赵波,李明,万平.小豆SSR分子标记遗传连锁图谱构建[J].中国农业科学,2013,17:3534-3544.
[26]Xia Jun Hong,Liu Feng,Zhu Ze Yuan,et al.A consensus linkage map of the grass carp(Ctenopharyngodon idella)based on microsatellites and SNPs.[J].BMC Genomics,2010,11.
[27]姚红伟,张立冬,孙金阳,刘霄霞.DNA分子标记技术概述[J].河北渔业,2010,07:42-46.
[28]宋宪亮,孙学振,张天真.偏分离及对植物遗传作图的影响[J].农业生物技术学报,2006,02:286-292.
[29]余渝.棉花种间群体配子重组率差异、偏分离研究与高密度分子标记遗传连锁图谱构建[D].华中农业大学,2010.
[30]M.Lorieux,B.Goffinet,X.Perrier,et al.Lanaud.Maximum-likelihood models for mapping genetic markers showing segregation distortion.1.Backcross populations[J].Theoretical and Applied Genetics,1995,901.
[31]S.Cloutier,M.Cappadocia,B.S.Landry.Analysis of RFLP mapping inaccuracy in Brassica napus L.[J].TAG Theoretical and Applied Genetics,1997,951-2.
Constructing a Genetic Linkage Map by SRAP and SSR Marker for Kenaf(Hibiscus cannabinus L.)
WU Yaolong,LI Defang,LI Jianjun,HUANG Siqi,LI Hui,TANG Huijuan,CHEN Anguo
(Institute of Bast Fiber Crops,Chinese Academy of Agricultural Sciences,Changsha 410205,China)
Construction of genetic linkage map for kenaf will lay good foundation for QTL mapping,QTL map-based cloning,superior gene screening and molecular marker-assisted breeding of essential agronomic traits.256 pairs of SRAP primers and 64 pairs of SSR primers of cotton were screened twice,150 F2derived from two kenaf varities,Thailand No.1 and F71,as mapping material.73 pairs of SRAP primers and 8 pairs of SSR primers were screened,and these primers are clearly visible and high polymorphic.A genetic linkage map with 128 DNA loci spanning 2155.43 cM was constructed.It contains 128 polymorphic makers scattered in 18 linkage groups with average interval of genetic linkage map 16.09 cM.In this paper,it is proved that SSR primers of cotton used for kenaf are available and genetic linkage map for kenaf has relatively high density and its markers are evenly distributed,which will be suitable for QTL mapping and QTL map-based cloning and etc.in the subsequent study.
genetic linkage map;kenaf;SRAP;SSR
S563.5
A
1671-3532(2016)06-0249-09
2016-06-28
国家麻类产业技术体系(CARS-19-E07)
武耀龙(1988-),男,在读研究生,专业方向作物遗传育种。E-mail:wuyaolong2012@126.com
*通讯作者:陈安国(1964-),男,研究员,主要从事一年生麻类遗传育种及新品种推广。Email:cagibfc@126.com
猜你喜欢
计算机应用与软件(2022年6期)2022-07-12
西南农业学报(2022年5期)2022-06-06
自然灾害学报(2022年2期)2022-05-10
中国麻业科学(2021年5期)2021-12-02
中国糖料(2021年3期)2021-07-13
中国学校体育(2021年10期)2021-04-26
中国麻业科学(2020年5期)2020-12-26
热带农业科技(2019年1期)2019-01-14
中国果业信息(2019年11期)2019-01-05
中国麻业科学(2018年6期)2018-04-09
