
基于氨基多羧酸螯合铁催化氧化硫化氢技术的研究进展
2016-12-23 09:15张世光
中北大学学报(自然科学版) 2016年6期
张世光, 罗 莹, 郭 芳
(1. 中北大学 超重力化工山西省重点实验室, 山西 太原 030051;2. 中北大学 山西省超重力化工工程技术研究中心, 山西 太原 030051)
基于氨基多羧酸螯合铁催化氧化硫化氢技术的研究进展
张世光1,2, 罗 莹1,2, 郭 芳1,2
(1. 中北大学 超重力化工山西省重点实验室, 山西 太原 030051;2. 中北大学 山西省超重力化工工程技术研究中心, 山西 太原 030051)
基于络合铁法脱硫的催化氧化技术用于脱除恶臭污染物H2S气体已成为能源加工业的研究热点. 本文综述了30年来国外脱硫过程中的络合化学、 反应机理及动力学等研究内容. 首先提出适宜螯合剂的平衡常数范围, 指出有氧供体的配体生成的Fe(Ⅲ)/Fe(Ⅱ)螯合剂的稳定性高; 其次认为吸收反应过程中各中间产物存在形态是pH值的函数, Fe(Ⅲ)通过羟基化的途径来增强反应活性, 中间物反应活性的差异与pH值、 螯合剂中配位基团的结构、 数量有关. 同时通过对螯合剂降解机理的比较分析指出配体被OH·氧化攻击的地方是在-亚甲基与羧酸盐基团之间, 亚甲基在乙烯桥中连接两个氮原子, 从而为如何抑制络合剂的降解提供了参考; 最后对我国络合铁法脱硫当前存在的问题及未来的研究方向进行了探讨.
螯合铁; 氧化还原; 硫化氢; 催化氧化; 降解
0 引 言
硫化氢(H2S)是一种具有恶臭气味的有毒气体, 来源于石油精炼、 纸浆制造、 天然气、 煤制气、 好氧和厌氧废水处理等工业加工过程. 到目前为止, 已有多种方法用于去除含硫化氢工业废气, 其中螯合铁脱硫工艺由于其对H2S的高脱硫率, 工艺操作弹性大, 被证明具有良好的经济潜力, 并且正逐步取代传统的湿法氧化还原装置[1]. 螯合铁脱硫过程是一个基于三价铁/二价铁氧化还原电对典型的液体氧化过程, 该方法实现了对Fe3+-Fe2+-Fe3+的循环利用, 对H2S的氧化还原过程如下[2]:
络合铁吸收硫化氢
HS(g)←→H2S(aq)
H2S(aq)+2Fe3+Ln-→S↓+2H++2Fe2+Ln-.
(1)
络合亚铁的再生
O2(g)←→O2(L)
O2(aq)+4Fe2+Ln-+2H2O→4Fe3+Ln-+4OH-,
(2)
目前络合铁法脱硫技术已实现工业化并取得显著成就的工艺有美国whee/abrator clean Air Systeems. Inc公司的Lo-Cat、 法国Le Gaz In tegral Enterprise公司的Sulfint、 美国Shell Oil和Dow chemical公司的Sulferox等工艺[3-4].
本文从近30年来国外对络合铁法研究中络合剂选择的基本原则入手, 分别探讨了脱硫过程中的络合化学、 反应机理及动力学等, 重点论述了Fe3+Ln-在脱硫吸收过程中反应中间活性基团等内容, 最后对不同络合剂的降解进行了比较分析并指出性能优异的络合剂的结构特征.
1 Fe(Ⅲ)/ Fe(Ⅱ)螯合剂的选择
铁的水解化学成为铁利用的障碍. 在络合铁法脱硫工艺吸收过程中, 一直存在着两个最大的问题: ① 螯合剂的选择, 是为了阻止不溶性铁化合物的生成且不影响铁的氧化还原能力; ② 配体的降解.
Fe(OH)3的Ksp=10-38.6, 当pH=8.5, CFe3+>10-22.9M时, 就会出现Fe(OH)3↓. 因此Fe(Ⅲ)螯合剂的稳定性必须高于Fe(OH)3↓(换言之, pM(-log[M])>22.9).
1960年国外学者发表了有关络合铁法脱硫的第一个专利, 20世纪70年代后人们逐步实验络合铁法脱硫的催化氧化技术的大规模工业化应用. 1976年美国ARI技术公司最先开发出Lo-Cat工艺[5-6], 该工艺相关专利指出络合铁水溶液包括至少两种铁络合剂, 其中一种铁络合剂为含有胺类物质的碱金属盐; 另一种铁络合剂为多羟基化合物组成的糖类化合物.
Martell A. E.[7]等通过对20余种螯合剂的研究发现Fe(Ⅲ)螯合剂条件平衡常数介于108~1024(pH=8.5, [HS-]=10-3M). 由于Fe (Ⅲ)是弱的硬酸, 易与含氧供体配体结合, 有氧供体的配体生成的Fe(Ⅲ)/Fe(Ⅱ)螯合剂的稳定性高, 有氧和氮供体的配体生成的Fe(Ⅲ)/Fe(Ⅱ)螯合剂的稳定性低, 如图 1 所示.

图 1 Fe2+螯合物的logK1与相应的Fe3+螯合物的logK1[7]Fig.1 Plot of logK1 for complexes of Fe2+ against logK1 for the corresponding complexes of Fe3+[7]
2 Fe3+L与H2S的反应机理与动力学
与大规模络合铁脱硫技术的工业发展相比, 相应理论研究起步较晚. 1991年Deberry[8]首次为络合铁法脱硫技术的理论研究提供了反应过程中的动力学数据, 到1997年时络合铁脱硫理论研究已有很大突破.
2.1 Fe(Ⅲ) 螯合剂的络合化学
长期以来, 众多学者一直在对络合铁法脱硫过程中Fe(Ⅲ)螯合剂存在的中间反应物形态、 种类以及氧化过程中具有反应活性的中间反应物进行不断的探索. 研究结果表明: Fe(Ⅲ)螯合剂在不同螯合体系里形成了多种中间反应物, 但CFe(III)是pH值的函数, 如图 2, 图 3 所示[9].

图 2 不同pH下Fe(Ⅲ)EDTA的浓度[9]Fig.2 Concentration of ferric chelates[9] EDTA in different pH

图 3 不同pH下Fe(Ⅲ)HEDTA的浓度[9]Fig.3 Concentration of ferric chelates[9] HEDTA in different pH
Gustafson和Martell[10]、 Schugar[11]、 Wilkins和Yelin[12]相继发现了Fe(Ⅲ)螯合剂存在的中间反应物形态、 种类及反应, 并认为酸催化造成了μ-氧化二聚体的分解.

(3)

(4)

(5)
反应(6)也认为通过下述途径进行

(6)
Philip和Brooks[13]首先发现Fe(Ⅲ)与HEDTA所形成螯合物在反应(1)里对pH有非常强的依赖性. 他们认为在高的pH值下, 由于Fe(Ⅲ)螯合物的羟基化和μ-氧桥键的形成造成对pH的依赖, 并推断在反应(4), (5)里羟基化后的各组分与H2S反应活性要远大于非羟基化组分, 这就表明具有μ-氧桥键的(Fe3+Ln-)2(O2-)没有反应活性, 但此组分可能参加了反应(1)而在反应(5), (6)里分解了. Asai[14-15](L=H2O)、 DeBerry[8]、 Wubs和Beenackers[16]、 Neyaglov[17](L=EDTA, HEDTA)、 Demmink[18](L=NTA)和Yao和Millero[19]等证实了Fe(Ⅲ)螯合物通过羟基化的途径来增强反应活性的现象.
如果配位基过量, 则发生下列反应

i=1,…,n.
(7)
2.2 反应机理
虽然络合铁法脱硫技术已实现商业化应用, 但相关动力学数据和机理研究数据较少, 表 1 对近30年来国外的研究结果进行了总结.

表 1 H2S和Fe(Ⅲ)螯合剂反应的研究
Neyaglov[17]、 Koch[21]和Lonergan[27]的机理研究表明一种亚稳定性的硫铁复合物的存在, 它的形成可表示为

(8)
Dos Santos和Stumm[22]、 Yao和Millero[19]在研究Fe(OH)3↓时也曾提及到类似的硫铁中间生成物.
虽然对于反应连续性的细节部分研究者们持有不同的观点, 但不论是涉及到H2S[17,27]还是内部电子转移其结果都表明Fe3+Ln-(SH)-实际参加了反应(9).
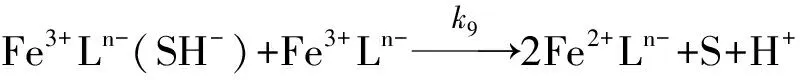
(9)
当提高pH值后, 可以观测到有高反应活性的反应发生, 这其中可能有羟基化后的组分
(10)

(11)
对于Fe(Ⅲ)HEDTA来说

(12)

(13)
所有文献研究报道在反应(1)里Fe(Ⅲ)和H2S是一级反应, 同时表明在反应区域里消耗了Fe(Ⅲ)螯合物, 因此反应(8)、 (10)、 (12)是速率控制步骤. Wubs和Beenackers[24]、 Demmink[18]都假定Fe3+Ln-(SH)-的浓度为稳态时, Fe3+Ln-、 Fe3+Ln-(OH)-、 Fe3+Ln-(OH)2有独立的反应路径, 这些离子浓度与CFe(Ⅲ)有关, 是CFe(Ⅲ)和pH的函数, 并进一步认为CFe(Ⅲ)较高时, 存在μ-氧化二聚体, 但没有与H2S反应, 也没有观测到CFe(Ⅲ)的二级反应[17,20].
当螯合剂为EDTA、 HEDTA时, 有学者[16,24]对高压和低压, 离子强度、 界面浓度梯度的研究推导出Fe(Ⅲ)螯合物的反应活性

(14)

2.3 反应动力学
最初的时候, Нодглов[28-29]等在实验的基础上提出的动力学方程式为

(15)
式中:k3=0.015±0.005 s-1,k8=650±501 mol-1·s-1,k2=160±51 mol-1,k11=0.43±0.05. 但作者未提供准确的活化能数据, 仅云活化能低, 在0~50 kJ·mol-1.
同时Нодглов[28-29]坚持认为添加元素硫对反应的影响表明反应有一诱导期, 显示其自动催化性质, 如图 4 所示.

有进行校正. 所有研究者都认为存在大量的一羟基二聚物和可能的二羟基化合物.

图 4 添加硫元素对反应的影响[28-29]Fig.4 Influence of added sulfur reaction[28-29]

反应物k1,1[9]/(m3·mol-1·s-1)k1,1[24]/(m3·mol-1·s-1)Ea[24]/(kJ·mol-1)Fe3+L4-EDTA(m0)≈0≈0Fe3+L4-EDTA(OH-)(m1)250~30014~21Fe3+L3-HEDTA(m0)≈0≈0Fe3+L3-HEDTA(OH-)(m1)1.51.8Fe3+L3-HEDTA(OH-)(m2)550~65013026.4±0.6
3 螯合剂降解机理
很长时间以来, 公开文献很少报导螯合剂降解机理的原因和机理, 只有Chen[7,30-31]的研究做了较为详细地分析.
3.1 其他学者的研究
一直以来研究者们[32-37]对于EDTA的水解存在很大争议, Ramunnas[38]研究发现当氨水浓度高时EDTA水解和氨解生成了UEDDA、 HEIDA和IDA, MIDA=MUEDDA+MHEIDA, 175 ℃时, 在稀释的氨溶液中, EDTA的降解一级反应常数为8.6×10-5·s-1, 无氨水时水解常数为4.2×10-5·s-1, 该反应的△H0=35 kcal/mol. 当用甲胺代替氨水时, 生成物UEDDA被N-(2-甲氧乙基)亚氨基二乙酸取代. 反应中随着丙氨基的生成水解的程度逐渐加剧, 由此提出了双分子SN2的反应机理, 伴随着亚氨基二乙酸的取代, C的乙烯桥毗邻了一个质子化了的EDTA上的N原子, 而EDTA的裂解正是因为醋酸基团复合物上的各个质子发生了化学位移.
McManus和Kin[39]、 Chen[30]、 Liu[40]提出在低pH值反应历程的过渡, 可能是敏感的不稳定复合物增强了降解性, 后者在过量NTA时观察到的更高降解率.
Simon[41-43]研究了Fe(Ⅲ) CDTA和Fe(Ⅲ)EDTA的降解, 研究表明: 高pH和高温是增加螯合物降解速度的主要因素. 高离子强度(也就是IC=0.5 mol/L) 也会促进降解. 在最苛刻的条件(T=55 ℃, pH=10,IC=0.5 mol/L)下, 高达40%的Fe(Ⅲ)CDTA和54%的Fe(Ⅲ) EDTA降解, 证明在碱性溶液中CDTA是防止铁沉淀的优良的螯合剂.
3.2 Chen的研究结果
Chen[7,30-31]的研究对NTA、 EDTA和HEDTA的相对降解速率进行了比较. 图 5 表明超过80%的EDTA在80 h内被氧化, IDA和EDDA作为氧化过程的中间体; HEDTA的氧化降解是相对较快的, 70 h后只剩下10%的原配体; NTA本身以及所有中间降解是缓慢而渐进的, 在100 h内约有80%的NTA被氧化.
Chen[7,30-31]认为络合剂的降解是因为再生过程中形成的过氧化物(或自由基)对络合剂的攻击造成的, 属于氧化降解. 在此过程中, 发生了一个芬顿反应类型的反应[44-45], 生成了H2O2, 进而与Fe2+反应生成了OH·. 这些配体被OH·氧化攻击的地方似乎是在α-亚甲基与羧酸盐基团之间, 同时亚甲基在乙烯桥中连接两个氮原子, 这就给如何抑制络合剂的降解提供了大的研究方向. 在Chen的研究中, 利用PBN作为捕获剂, 通过电子自旋共振谱检测出羟基的羟基化作用生成OH·, 表明在氧化还原反应中有OH·生成, 如图 6 和图 7 所示.

图 5 Fe(III)-NTA、 Fe(III)- EDTA、 Fe(III)-HEDTA的降解[31]Fig.5 Degradation of Fe(III)-NTA, Fe(III)-EDTA, and Fe(III)-HEDTA [31]

图 6 Fe(II)-NTA和 H2O2的反应产物在pH=8.5的电子自旋共振谱Fig.6 ESR spectrum of the reaction product of Fe(II)-NTA with H2O2 at pH=8.5

图 7 Fe(III)-NTA和H2S、 空气混合气体的反应产物在pH=8.5的电子自旋共振谱Fig.7 ESR spectrum of the reaction product of Fe(III)-NTA with H2S and air at pH=8.5
4 结论与展望
在整个脱硫工艺的过程中, 可溶性铁离子与螯合剂的结合、 释放的动力学、 热力学、 作用机理等问题对于了解络合铁的化学活性十分重要, 但由于螯合剂结构中存在两个不同的结合位点, 目前尚未能区分铁是在N端结合抑或C端结合.
我国要想尽快提升络合铁法脱硫技术的研究与应用水平, 可以从以下三个方面着手:
1) 对于理论研究, 可通过复制模型进行研究: ① 所有离子物种的局部浓度梯度; ② 界面附近Fe(Ⅲ)螯合物互变产生的化学平衡和由H2S造成更多活性物种的消耗; ③ 离子扩散, 包括静态和动态电中性条件; ④ 反应(1)的反应动力学、 反应机理、 包括铁螯合物的络合化学.
2) 传统络合剂降解位置是醋酸侧链或乙烯桥的亚甲基, 今后更应关注不易被OH·攻击的亚甲基的配体上, 如吡啶二羧酸类物质、 羟基化芳环配体, 尽可能地消除羟基自由基, 因为它已被证实在络合铁氧化降解中作为活性氧化剂.
3) 开发绿色螯合剂以及各种改进的氨基多羧酸型, 最有希望的替代品是易生物降解的氨基多羧酸型螯合剂如GLDA、 EDG(或HEIDA)、 MGDA、 IDS和EDDS, 其中GLDA因其绿色属性和可接受性而备受关注.
[1]Zhai Linfeng, Hu Lili, Sun Min. Understanding the catalyst regeneration kinetics in the chelated iron dehydrosulfurization process: a model in terms of Fe(II) speciation[J]. Industrial and Engineering Chemistry Research, 2015, 54: 6430-6437.
[2]Wubs H J, Beenackers A A C M. Kinetics of the oxidation of ferrous chelates of EDTA and HEDTA in aqueous solution[J]. Industrial and Engineering Chemistry Research, 1993, 32(11): 2580-2594.
[3]Stepova K V, Maquarrie D J, Krip I M. Modified bentonites as adsorbents of hydrogen sulfide gases[J]. Applied Clay Science, 2009, 42(3): 625-628.
[4]Nassar N N, Husein M M, Pereira-Almao P. Ultradispersed particles in heavy oil: Part II, sorption of H2S(g)[J]. Fuel Processing Technology, 2010, 91(2): 169-174.
[5]Thompson R B. Composition for catalytic removal of hydrogen sulfide from gases: U.S., 4218342[P]. 1980-08-19.
[6]Thompson R B. Catalytic removal of hydrogen sulfide from gases: U.S., 4189462[P]. 1980-02-19.
[7]Martell A E, Motekaitis R J, Chen D, et al. Selection of new Fe (III)/Fe (II) chelating agents as catalysts for the oxidation of hydrogen sulfide to sulfur by air[J]. Canadian Journal of Chemistry, 1996, 74(10): 1872-1879.
[8]DeBerry D W, Petrinec B, Trofe T. New insights from investigation of fundamental mechanisms of liquid redox chemistry[C]. Liquid Sulfur Recovery Conference, 1991.
[9]Demmink J F, Beenackers A. Gas desulfurization with ferric chelates of EDTA and HEDTA: new model for the oxidative absorption of hydrogen sulfide[J]. Industrial and Engineering Chemistry Research, 1998, 37(4): 1444-1453.
[10]Gustafson R L, Martell A E. Hydrolytic tendencies of ferric chelates[J]. The Journal of Physical Chemistry, 1963, 67(3): 576-582.
[11]Schugar H J, Hubbard A T, Anson F C, et al. Electro chemical and spectral studies of dimeric iron (III) complexes[J]. Journal of the American Chemical Society, 1969, 91(1): 71-77.
[12]Wilkins R G, Yelin R E. Kinetics of monomer-dimer interconversion of iron (III) ethylenediaminetetraacetate and related chelates[J]. Inorganic Chemistry, 1969, 8(7): 1470-1473.
[13]Philip C V, Brooks D W. Iron(III)chelate complexes of hydrogen sulfide and mercaptans in aqueous solution[J]. Inorganic Chemistry, 1974, 13(2): 384-386.
[14]Asai S, Konishi Y, Yabu T. Kinetics of absorption of hydrogen sulfide into aqueous ferric sulfate solutions[J]. AIChE Journal, 1990, 36(9): 1331-1338.
[15]Asai S, Nakamura H, Aikawa H. Absorption of hydrogen sulfide into aqueous ferric chloride solutions[J]. Journal of Chemical Engineering of Japan, 1997, 30(3): 500-506.
[16]Wubs H J. Application of iron chelates in hydrodesulphurisation[D]. University of Groningen, 1994.
[17]Neyaglov A A, Digurov N G, Bukharkina T V, et al. Kinetics and reaction mechanism of the liquid-phase oxidation of hydrogen sulfide by a chelate complex of trivalent iron (Fe3+EDTA-)[J]. Kinetics and Catalysis, 1991, 32(3): 485-489.
[18]Demmink J F, Wubs H J, Beenackers A A C M. Oxidative absorption of hydrogen sulfide by a solution of ferric nitrilotriacetic acid complex in a cocurrent down flow column packed with SMV-4 static mixers[J]. Industrial and Engineering Chemistry Research, 1994, 33(12): 2989-2995.
[19]Yao W, Millero F J. Oxidation of hydrogen sulfide by hydrous Fe (III) oxides in seawater[J]. Marine Chemistry, 1996, 52(1): 1-16.
[20]Neumann D W, Lynn S. Oxidative absorption of H2S and O2by iron chelate solutions[J]. AIChE journal, 1984, 30(1): 62-69.
[21]Koch S, Ackermann G, Schüller K. Über ternäre Komplexe von Eisen (III) mit Aminopolycar-bonsäuren und Sulfid[J]. Zeitschrift für Chemie, 1986, 26(9): 339-339.
[22]Dos Santos A M, Stumm W. Reductive dissolution of iron (III)(hydr) oxides by hydrogen sulfide[J]. Langmuir, 1992, 8(6): 1671-1675.
[23]DeBerry D W. Rates and mechanisms of reaction of hydrogen sulfide with Iron chelates[R]. U.S.: Gas Research Institute, 1993.
[24]Wubs H J, Beenackers A A C M. Kinetics of H2S absorption into aqueous ferric solutions of EDTA and HEDTA[J]. AIChE Journal, 1994, 40(3): 433-444.
[25]Giles T W R, Smith J W, Tombalaikan A S. Kinetic study of the catalytic oxidation of hydrogen sulphide in industrial waste streams[J]. Waste Process. Recycl, 1994: 229-239.
[26]Clarke E T, Solouki T, Russell D H, et al. Transformation of polysulfidic sulfur to elemental sulfur in a chelated iron, hydrogen sulfide oxidation process[J]. Analytica Chimica Acta, 1994, 299(1): 97-111.
[27]Lonergan M C, Lieberman M, Lewis N S. Mechanistic studies of and redox sensors for the liquid redox process[C]. GRI Sulfur Recovery Conference, Austin, TX. Gas Research Institute: Chicago, IL, 1995.
[28]Неяглов А А, Н. Г Дигуров, Г. В. Бухаркина. Новые катапические системы на основе водокнистых ионитов. лр. Кцнет. Кат[J]. 1991, 32(4): 963-967.
[29]Неяглов А А, Н. Г Дигуров, Г. В. В. Бухаркина. Очистка широкои фракции легких угпёъоаороаов газового конаёнсата месторожаения от сернистых соединении. лр. Кцнет. Кат[J]. 1991, 32(3): 541-547.
[30]Chen D, Motekaitis R J, Martell A E, et al. Oxidation of H2S to S by air with Fe (III)-NTA as a catalyst: catalyst degradation[J]. Canadian Journal of Chemistry, 1993, 71(9): 1524-1531.
[31]Chen D, Martell A E, McManus D. Studies on the mechanism of chelate degradation in iron-based, liquid redox H2S removal processes[J]. Canadian Journal of Chemistry, 1995, 73(2): 264-274.
[32]Martell A E, Motekaitis R J, Fried A R, et al. Thermal decomposition of EDTA, NTA, and nitrilotrimethylenephosphonic acid in aqueous solution[J]. Canadian Journal of Chemistry, 1975, 53(22): 3471-3476.
[33]Venezky D L, Moniz W B. Thermal Stability of Ethylenedinitrilo Tetraacetic Acid and its Salts. Part 2. Rate of Decomposition in Aqueous Solution Determined by NMR Techniques[R]. Washington DC: Naval Research Lab, 1968.
[34]Venezky D L, Moniz W B. Nuclear magnetic resonance study of the thermal decomposition of ethylenedinitrilotetraacetic acid and its salts in aqueous solutions[J]. Analytical Chemistry, 1969, 41(1): 11-16.
[35]Osborn D, Wilson J S, Fried A R, et al. Final Report of Contract G-9-2-D[J]. American Society of Mechanical Engineers, 1973, 85-92.
[36]Martell A E, Fried J A R. Structure and conformation of thorium (IV) complexes of diethylenetriaminepentaacetic acid in aqueous solution[J]. Journal of the American Chemical Society, 1971, 93(19): 4695-4700.
[37]Venezky D L, Rudzinski W E. Determination of ethylenediaminetetraacetic acid in boiler water by liquid chromatography[J]. Analytical Chemistry, 1984, 56(2): 315-317.
[38]McLendon G, Motekaitis R J, Martell A E. Kinetics and mechanism of mu.-oxo dimer formation by ethylenediaminetetraacetatatoiron (III)[FeEDTA][J]. Inorganic Chemistry, 1976, 15(9): 2306-2308.
[39]McManus D, Kin F R. Hydrogen sulfide removal: U.S., 4622212[P]. 1986-11-11.
[40]Liu X, Sawyer D T, Bedell S A, et al. Ligand degradation in the iron/dioxygen-induced dehydrogenation of H2S[C]. Seventh Sulfur Recovery Conference, Austin, TX., 1995: 24.
[41]Piche S, Ribeiro N, Bacaoui A, et al. Assessment of a redox alkaline/iron-chelate absorption process for the removal of dilute hydrogen sulfide in air emissions[J]. Chemical Engineering Science, 2005, 60(22): 6452-6461.
[42]Piché S, Larachi F. Dynamics of pH on the oxidation of HS-with iron (III) chelates in anoxic conditions[J]. Chemical Engineering Science, 2006, 61(23): 7673-7683.
[43]Piché S, Larachi F. Kinetic effect of electrolytes on the oligomerization of hydrosulfide into polysulfides and colloidal sulfur with iron(III) trans-1, 2-diaminocyclohexanetetraacetic acid in anoxic aqueous solutions[J]. Chemical Engineering Science, 2006, 61(21): 7171-7176.
[44]Metelitsa D I. Mechanisms of the hydroxylation of aromatic compounds[J]. Russian Chemical Reviews, 1971, 40(7): 563-580.
[45]Vysotskaya N A. The reactivities of·OH,·O-, and HO2· radicals and Oxygen atoms in aqueous solutions of aromatic compounds[J]. Russian Chemical Reviews, 1973, 42(10): 851-856.
Study on Catalyst Oxidetion H2S with Aminopolycarboxylic Chelating Iron
ZHANG Shi-guang1,2, LUO Ying1,2, GUO Fang1,2
(1. Shanxi Province Key laboratory of High Gravity Chemical Engineering, North University of China, Taiyuan 030051, China; 2. Research Center of Shanxi Province for High Gravity Chemical Engineering and Technology, North University of China, Taiyuan 030051, China)
Gas desulfurization processes based on iron chelate chemistry as a catalytic oxidization technology for the removal of H2S in energy processing industries has received increasing attention. Complex chemistry, mechanism and dynamics of desulfurization processes based on iron chelate are reviewed in recent 30 years. Firstly, an unexpected result of this study is that favorable redox reactions are associated with conditional constants that vary over a wide range of numerical values, ligands that form suitable iron complexes as catalysts have oxygen donors. Then, it is the hydroxyl radical that has ability to oxidation Fe(Ⅲ) chelates whereas ferryl has not. It is seen that pH has significant impact on the ferric chelate complexs on the reactive absorption rate of H2S, and the phenomenon of increasing reactivity of ferric chelates with hydroxylation is shown too. The observation indicates that pH, the number and structure of coordinating groups on chelates are related to the difference of intermediate reactivity. Furthermore, the intermediates and products of the oxidative degradation of each chelating agent are described and compared. During the reoxidation of the ferrous formed and which in turn generates the hydroxyl radical by reaction with ferrous ion. The site of oxidative attack by the hydroxyl radical on these ligands seems to be the -CH2- groups a to the carboxylate groups, as well as the -CH2- groups in the ethylene bridges between the nitrogen. Lastly, the current problems as well as the corresponding research directions are discussed.
iron chelates; redox; hydrogen sulfide; redox catalysts; degradation
1673-3193(2016)06-0625-08
2016-06-08
国家自然科学基金资助项目(21376229)
张世光(1981-), 男, 讲师, 博士生, 主要从事超重力技术及废水处理工艺的研究.
TQ028.8
A
10.3969/j.issn.1673-3193.2016.06.013
猜你喜欢
环境科学研究(2022年10期)2022-10-19
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
中国土壤与肥料(2021年5期)2021-12-02
中国农业气象(2021年12期)2021-11-30
无机化学学报(2020年7期)2020-07-20
河北果树(2020年2期)2020-05-25
渤海大学学报(自然科学版)(2020年3期)2020-02-02
中成药(2017年9期)2017-12-19
中成药(2017年9期)2017-12-19
中成药(2017年7期)2017-11-22
