
特发性肺纤维化1例
2016-12-22 02:29:36闫利娟秦芸芸
外科研究与新技术 2016年2期
闫利娟,秦芸芸
1.洛阳市第二中医院病理科,洛阳471003;2.上海兰卫临床医学检验所病理室,上海200335
·综述·
特发性肺纤维化1例
闫利娟1,秦芸芸2
1.洛阳市第二中医院病理科,洛阳471003;2.上海兰卫临床医学检验所病理室,上海200335
目的 探讨特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)的临床和病理特点。方法 对1例IPF进行临床-影像-病理诊断,并探讨其临床病理特点。结果 患者为老年男性,病情呈进行性加重,肺功能显示限制性通气障碍和弥散功能降低,影像和病理具有普通型间质性肺炎的特点。结论 IPF的病理诊断需要密切结合临床和影像学。
特发性肺纤维化;特发性间质性肺炎;诊断
患者男性,70岁,进行性气急2年,活动后加剧。2007年12月外院肺功能检查,显示“限制性通气障碍和弥散功能降低”,行胸部CT检查,表现为以胸膜下和下叶分布为主的网状影、蜂窝肺(图1)。近一年来症状呈进行性加重,活动后气急,需深大呼吸后缓解,遂于2009年8月1日入院治疗。查体:体温37℃,脉搏120次/m in,呼吸23次/m in,血压110/60mmHg。患者呼吸急促,体型消瘦,两肺呼吸运动、呼吸动度及触觉语颤对称,双肺呼吸音粗,两肺底可闻及Velcro音,可见杵状指。血常规:白细胞计数10.5×109/L,中性粒细胞82.1%,C-反应蛋白(CRP)53 mg/L。血气分析:pH值7.43,PaO280 mmHg,SaO296%,PaCO239mmHg。入院后完善相关检查,全麻下行胸腔镜下肺活检术。肺活检病理组织学检查,病变呈斑片状分布,轻重不一、新旧病变交杂,肺泡结构破坏,间质炎症性改变,纤维化伴蜂窝肺形成,可见纤维母细胞灶形成(图2)。病理诊断为“普通型间质性肺炎(usual interstitial pneumonia,UIP)”。经临床-影像-病理诊断为特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)。诊断后遂给予糖皮质激素治疗,疗效不明显,患者一个月后死于呼吸衰竭。

图1 胸部CT显示,两肺片状、以基底部为主的网状阴影和磨玻璃影,两肺胸膜下有蜂窝样改变Fig.1 High-resolution CT images demonstrated basal predom inant,peripheralpredom inant reticular abnorm ality and ground glassabnorm ality w ith subp leural honeycom bing
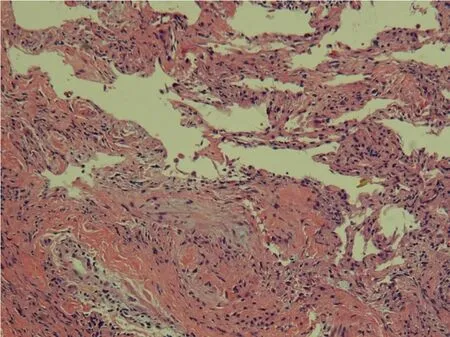
图2 光镜观察,肺间质纤维组织增生,肺泡结构改建,有纤维母细胞灶形成(中)(HE×100)Fig.2 Lightm icroscopic exam ination dem onstrated fibrosisextending to the pu lm onary interstitium,alveolar structural rem odeling and the regionsof fibrob last focus (m idd le)(HE×100)
2 讨论
IPF是一种原因不明、局限于肺部的弥漫性、进行性慢性纤维性间质性肺炎,是特发性间质性肺炎(idiopathic interstitial pneumonias,IIPs)的主要类型(占65%左右),目前无特效治疗,预后与肺癌类似[1],在病理和影像学上表现为UIP[2]。IPF的诊断一直是临床和病理的难点[3]。2002年美国胸科学会(ATS)和欧洲呼吸学会(ERS)发表IIPs分类和诊断标准国际共识,即IIPs的ATS/ERS分类[4]。该分类强调,IIPs各型的诊断除临床和影像学资料外,明确诊断依赖于电视胸腔镜手术/开胸肺活检,但最后的病理诊断应密切联系临床资料和影像学,即临床-影像-病理 诊断(clinico-radiologic-patho-logic diagnosis,CRP诊断),单独由临床医师、放射科医师或病理科医师作出诊断都有可能是片面的,应尽可能进行CRP诊断。
IPF发病年龄常在50~70岁之间,约2/3的患者发病时超过60岁,本例就是一位70岁的老年患者。临床表现为不明原因的干咳、呼吸困难等,可闻及吸气性爆裂音,以双肺底部最为明显,可见杵状指[3]。肺功能检查显示中至重度限制性通气功能障碍和/或弥散功能障碍。对糖皮质激素反应差,预后差。影像学检查对IPF的诊断具有重要价值,包括胸部X线片和计算机断层扫描(CT)或高分辨CT (HRCT)。X线表现:病变主要分布于两下肺中外带,肺中央也可受累及,但从胸膜下至肺门,从肺底至肺尖病变逐渐减轻。而CT特别是HRCT能更为准确的评估IPF的肺部病变,可用于诊断、鉴别诊断和判断IPF活动性、严重程度和对激素的反应等。其表现为:(1)磨玻璃样密度影;(2)网状改变;(3)蜂窝肺;(4)胸膜下间质纤维化;(5)肺气肿;(6)牵拉支气管扩张,以上特点为IPF的基本征象而非特有,诊断时需与其他原因引起的间质纤维化鉴别[1]。IPF的病理学特点是:主要累及两肺胸膜下及肺实质,以双下肺为重;病变进展不一致,间质的炎症、纤维化和蜂窝变,不同时相的病变共存(新旧病变交杂分布),病变间可见正常肺组织;除了老病灶(胶原沉积的瘢痕灶)外,还有成纤维细胞灶,后者是由多量成纤维母细胞组成,具有黏液基质的背景,位于肺间质,突向被覆呼吸上皮的腔面;在纤维化区和蜂窝肺区可见有呼吸性细支气管、肺泡管以及重建的囊壁内有大量增生之平滑肌束,形成所谓的“肌硬化”[2]。本例从临床表现和影像学特点都提示IPF的可能;但是,由于发病少见,临床缺乏对该病的认识,最后为了明确诊断,进行了胸腔镜下肺活检病理诊断。病理学上显示了UIP的形态特点,通过CRP诊断为IPF,从而对该病的认识进一步提高。
2011版IPF诊治指南[1]指出,UIP的HRCT特点可作为独立的IPF诊断手段,具备UIP典型HRCT表现者不必行病理活检;因为一些研究证实,HRCT诊断UIP准确性可达到90%~100%,因此新指南废除了2000ATS/ERS共识中提出的IPF的主要和次要诊断标准。当然,影像学的诊断也是建立在临床经验的基础上。因此应仔细询问病史,密切注意临床表现和体征的变化,监测影像学改变,从而综合临床-影像-病理三方面的特点做出最后的诊断。
近年来,在临床接诊的患者中,肺纤维化的患者正逐年增多,尤其是IPF,但目前对其尚无确实、有效的方法。本例也显示皮质激素对IPF无效。因此寻找有效的治疗药物是目前研究的热点之一。ATS/ERS推荐使用糖皮质激素加硫唑嘌呤或环磷酰胺进行抗炎治疗,其中糖皮质激素是应用最广的一种治疗药物,但其对慢性期的肺纤维化作用较弱,远期疗效不佳。随着对本病发病机制和病理生理变化的深入研究和分子生物学技术的应用,治疗上有了新的进展,如抗纤维化治疗、抗氧化治疗,细胞因子治疗,基因治疗等。而肺移植是肺部疾病终末期的重要治疗手段之一,它对于患者的预后,尤其是间质性肺疾病有一定的改善[2],但供体的缺乏及高额的费用限制了肺移植的应用。
[1]Raghu G,Collard HR,Egan JJ,et al.ATS/ERS/JRS/ALAT comm ittee on idiopathic pulmonary fibrosis.an official ATS/ ERS/JRS/ALATstatement:idiopathicpulmonaryfibrosis:evidence-based guidelines for diagnosis and management[J]. Am JRespir CritCareM ed,2011,183(6):788-824.
[2]Travis WD,Costabel U,Hansell DM,et al.An Official AmericanThoracicSociety/EuropeanRespiratorySociety Statement:updateoftheInternationalMultidisciplinary Classification of the Idiopathic Interstitial Pneumonias[J].Am JRespir CritCareM ed,2013,188(6):733-748.
[3]American Thoracic Society,European Respiratory Society. AmericanThoracicSociety/EuropeanRespiratorySociety idiopathicpulmonaryfibrosis:diagnosisandtreatment. International consensus statement[J].Am J Respir Crit Care M ed,2000,161(2 Pt1):646-664.
[4]American Thoracic Society,European Respiratory Society. AmericanThoracicSociety/EuropeanRespiratorySociety international multidisciplinary consensus classification of the idiopathic interstitial pneumonias[J].Am J Respir Crit Care M ed,2002,165(2):277-304.
Idiopathic pulmonary fibrosis:a case report
YAN Lijuan1,QIN Yunyun2
1.DepartmentofPathology,Luoyang the Second HospitalofTraditionalChinese Medicine,Luoyang 471003,China;2.DepartmentofPathology,Shanghai Labway Clinical Laboratory Co.,Shanghai 200335,China
Ob jective To investigate the clinical and pathological features of idiopathic pulmonary fibrosis(IPF). M ethods One patient w ith IPF was diagnosed by integrating clinical,imaging and pathological information,and the clinicopathological characteristics of this case were analyzed.Results The patient described in the present case was a senior male w ith history of progressive dyspnea.The pulmonary function test revealed severe restrictive ventilatory impairment,and the diffusing capacity for carbon monoxide was reduced.The imaging and pathological exam inations demonstrated a pattern of usual interstitial pneumonia.ConclusionThe pathological diagnosis of IPF should be determ ined in consideration of clinical featuresand imaging findings.
Idiopathic pulmonary fibrosis;Idiopathic interstitialpneumonia;Diagnosis
R365
A
2095-378X(2016)02-0115-03
10.3969/j.issn.2095-378X.2016.02.013
闫利娟(1972—),女,大学本科,主治医师,从事临床病理诊断工作
秦芸芸,电子信箱:493408721@qq.com
(2016-01-28)
猜你喜欢
中老年保健(2022年2期)2022-11-25 23:46:31
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
昆明医科大学学报(2020年12期)2021-01-26 00:43:54
家庭医学(下半月)(2020年2期)2020-05-11 02:07:22
国际呼吸杂志(2019年21期)2019-11-25 09:52:20
中国中医药信息杂志(2016年7期)2016-12-01 06:07:52
中国卫生标准管理(2015年7期)2016-01-15 03:58:41
医学研究杂志(2015年8期)2015-06-22 14:00:57
天津医科大学学报(2015年3期)2015-06-05 12:21:49
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:43
