
生物法还原高浓度高氯酸盐动力学及反应条件的优化
2016-12-22 08:42吴敏王帅锋高乃云朱延平李硕朱思瑞
中南大学学报(自然科学版) 2016年11期
吴敏,王帅锋,高乃云,朱延平,李硕,朱思瑞
生物法还原高浓度高氯酸盐动力学及反应条件的优化
吴敏,王帅锋,高乃云,朱延平,李硕,朱思瑞
(同济大学污染控制与资源化研究国家重点实验室,上海,200092)
在高质量浓度高氯酸盐ClO4−(1 500 mg/L) 的条件下,驯化得到能够处理高质量浓度ClO4−废水的异养厌氧高氯酸盐还原菌群。通过摇床实验,用控制变量法探究初始ClO4−质量浓度(0~1 500 mg/L)、乙酸钠质量浓度(0~11.83 g/L)、pH(5.55~9.00)、温度(15~40℃)、共存离子SO42−质量浓度(0~26.0 g/L)和NO3−质量浓度(0~3.9 g/L) 对高质量浓度ClO4−生物还原性能的影响,确定最佳的反应条件。结果表明:最大比去除速率max和半饱和常数s分别为0.89 (mg·mg)/h和141.6 mg/L,驯化的高氯酸盐还原菌群具有处理高质量浓度ClO4−的潜力;在初始ClO4−质量浓度为1 300 mg/L时,乙酸钠的最佳投加量、最适pH和最适温度分别为6.5 g/L,35℃和6.85,常见共存离子SO42−和NO3−都会不同程度的抑制ClO4−的还原,且NO3−对ClO4−还原的抑制作用远高于SO42−。驯化污泥中,sp. 是ClO4−还原微生物的优势种属,占总菌的69.32%。
高氯酸盐;高质量浓度;高氯酸盐还原菌;动力学;优化
高氯酸盐(ClO4−)是环境中的主要无机污染物之一,广泛存在于地表水、地下水和土壤中,其高度的化学稳定性和良好的迁移性[1−3],使其成为环境污染治理的一大挑战。环境中的高氯酸盐主要来源于农业、工业和军事,在这些领域中,高氯酸盐主要作为化肥的原料,皮革加工、橡胶制造、涂料和润滑油生产等的添加剂以及火箭、导弹、焰火等的固体氧化剂[4]。饮用水中的高氯酸盐主要通过干扰甲状腺对碘的利用,使甲状腺激素的合成受到抑制,从而威胁人体健康[5]。美国环保署(US EPA)建议将饮用水中的高氯酸盐质量浓度标准定为15 μg/L[6],国内尚无相关的标准可参考。目前,高氯酸盐还原技术主要分为生物还原、化学还原和电化学还原。其中,利用异养高氯酸盐还原菌(DPRB)将ClO4−还原成Cl−的生物还原方法是去除高氯酸盐的一种廉价高效的处理技术,并且已经在实际工程中利用,取得良好的效果[7]。DPRB还原ClO4−的过程为ClO4−→ClO3−→ClO2−→Cl−+O2[8]。江毅[9]研究了不同有机碳源对DPRB还原速率的影响,发现丙酮酸钠和醋酸钠是DPRB最有效电子供体。WANG等[10]研究了生物法去除饮用水中的高氯酸盐的动力学和pH的影响,指出DPRB的pH作用范围为5~9。DUGAN等[11]通过中试试验证明了温度对DPRB的活性有显著的影响,并且在高于10℃的温度下,DPRB都能将50 μg/L左右的ClO4−还原到质量浓度2 μg/L的检测限以下。WU等[12]证明了微生物还原法可以在24 h以内将初始质量浓度为50 mg/L的ClO4−还原到检测限以下。但是,目前生物法去除高氯酸盐的研究对象主要为饮用水或地下水,这些水体中的ClO4−质量浓度较低,大都小于150 mg/L[13],而对生物法处理高浓度高氯酸盐废水的研究较少。事实上,一些地下水、工业和军事设施废水中的ClO4−质量浓度很高,在160~3 000 mg/L之 间[14],此外采用离子交换树脂处理高氯酸盐的饮用水时,也会产生高浓度的高氯酸盐洗脱液需要处理[15]。因此,系统研究高浓度高氯酸盐的生物还原具有重要意义。本实验驯化并富集了高浓度异养高氯酸盐还原菌,并通过高氯酸盐还原的序批式实验(SBR),研究了DPRB还原高浓度高氯酸盐的反应动力学,同时还分析了高氯酸盐浓度、碳源投加量、初始pH、反应温度、共存离子SO42−和NO3−对还原过程的影响,为生物还原高浓度高氯酸盐废水的实际应用提供指导和借鉴。
1 材料和方法
1.1 实验材料
本实验采用的驯化接种泥取自上海曲阳污水处理厂的厌氧硝化污泥。
培养液组成如表1所示。
碳源为无水乙酸钠(CH3COONa),纯度(质量分数)≥99.0%,购自国药集团有限公司;高氯酸钠(NaClO4∙H2O)的纯度≥99.0%,购自sigma-aldrich;用于厌氧处理的气体为高纯氮(N2体积分数≥99.999%),购自上海祥堃特种气体有限公司。
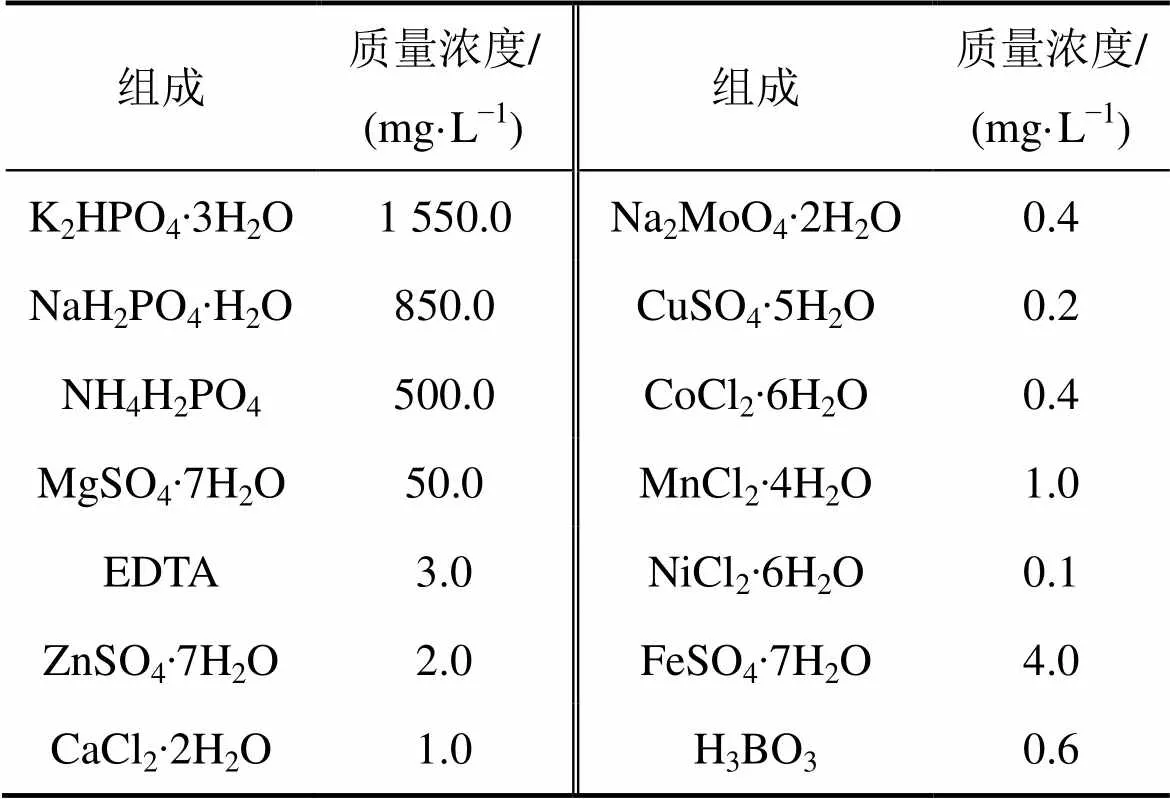
表1 培养液化学成分
注:所选化学试剂均为分析纯及以上,购自国药集团有限公司,培养液pH控制在7左右(用磷酸盐缓冲液进行调节)。
1.2 实验方法
1.2.1 高氯酸盐混合菌的培养与驯化
在9个250 mL的培养瓶中依次加入一定量的接种污泥、1.30 g乙酸钠、0.423 6 g高氯酸钠(NaClO4∙H2O)以及新鲜培养液,使每个培养瓶中的混合液体积、接种泥质量浓度、乙酸钠质量浓度和ClO4−质量浓度分别为200 mL,1 000 mg/L,6.50 g/L和1 500 mg/L,持续曝N2约5 min以赶走反应器中的O2(溶解氧DO质量浓度≤0.2 mg/L),瓶口用橡胶塞密封,最后放入温度和转速分别为35℃和150 r/min的恒温摇床中培养。每隔24 h,将培养瓶静置1.5 h,缓缓将上层清液倾倒出,补充一定量的乙酸钠和高氯酸钠,重新加入培养液至200 mL,重复以上操作。连续培养约60 d后,检测到ClO4−的还原稳定高效,表示驯化完成。
1.2.2 高氯酸盐还原的序批实验
ClO4−还原的序批实验(SBR)在250 mL的培养瓶中进行。在250 mL的培养瓶中加入驯化好的高氯酸盐混合菌、特定质量浓度的高氯酸钠和乙酸钠,然后加入培养液,使混合液的总体积和污泥质量浓度分别为200 mL和100 mg/L,经曝气后密封,放入恒温摇床中培养。实验过程如表2所示,即用控制变量法探究初始ClO4−质量浓度(0~1 500 mg/L)、乙酸钠质量浓度(0~11.83 g/L)、pH(5.55~9.00)、温度(15~40℃)、共存离子SO42−(0~26.0 g/L)和NO3−(0~3.9 g/L)对高质量浓度ClO4−生物还原性能的影响。
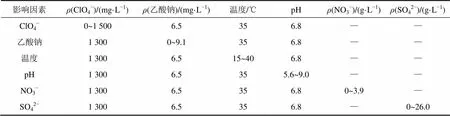
表2 序批式实验过程
注:“—”表示不投加,以上每组实验均设置3个平行试样。
1.3 分析方法
采用美国戴安ICS−1000型离子色谱仪对ClO4−质量浓度进行检测,色谱柱为IonPacAS20(4×250 mm) 阴离子分离柱,IonPacAG20(4×50 mm) 阴离子保护柱,分析软件采用Dionex Chromeleon。测定ClO4−的色谱参数:泵流速1.0 mL/min,淋洗液KOH质量浓度35 mmol/L,抑制电流100 mA,进样体积100 μL。样品采集后,首先在离心机中以3 000 r/min转速离心3 min,取上层清液经0.22 μm微孔滤膜过滤,并稀释一定的倍数使ClO4−离子质量浓度在检测范围内。
混合液pH由Mettler Toledo pH计测定,DO用WTW溶氧仪Oxi 3210测定,污泥质量浓度采用质量法测定。
高质量浓度高氯酸盐还原菌群的鉴定采用第2代高通量测序技术,由上海美吉生物医药科技有限公司测定。
2 结果与讨论
2.1 高氯酸盐还原动力学分析
实验中,初始ClO4−质量浓度分别取0,50,100,300,500,700,900,1 100,1 300和1 500 mg/L,得出其对ClO4−厌氧还原速率的影响如图1(a)所示。表明ClO4−的还原速率随着ClO4−质量浓度增大而增大,当ClO4−质量浓度达到1 500 mg/L时,ClO4−的还原速率趋于平稳。

(a) 不同初始ClO4−质量浓度对ClO4−还原速率的影响;(b) Monod方程的拟合曲线
DPRB增殖速率与底物ClO4−质量浓度之间关系用Monod动态模型分析,如下式所示:

将式(1)作如下的线性变形:
(2)
式中:为ClO4−还原速率,mg/(L·h);为ClO4−质量浓度,mg/L);s为半饱和常数,mg/L;为微生物质量浓度,mg/L;max为ClO4−的最大比去除速率,(mg·mg)/h;为反应时间,h。Monod方程拟合结果如图1(b)所示,拟合曲线的相关系数2为0.982 6,拟合效果较好,可表示为下述方程:
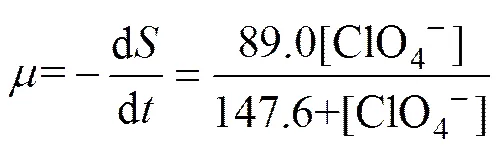
由拟合方程可知:max和s分别等于0.89 (mg·mg)/h和147.6 mg/L,与SEONG等[16]研究生物法处理高质量浓度高氯酸盐废水动力学,得出的max和s(分别为0.67 (mg·mg)/h和193.8 mg/L) 相近,都远远高于王蕊等[17]的生物法还原低质量浓度高氯酸盐地下水动力学中max和s(分别为0.45 (mg·mg)/h和12.6 mg/L)的研究结果。实验中较大max说明了整个反应过程的效率高,ClO4−最大反应速率快,而较大的s表明酶与基质的亲和力较小,增大ClO4−的质量浓度能够显著提高其还原速率[10],与图1(a)中的结果一致。以上表明本研究中在高质量浓度ClO4−下驯化得到的混合菌可以应用于高质量浓度ClO4−(100~1 500 mg/L)的还原。
2.2 碳源质量浓度对ClO4−还原的影响
CH3COO−还原ClO4−的反应过程如下[18]:
CH3COO−+H++ClO4−=2CO2+ Cl−+2H2O (4)
由反应式(4)可知:在初始ClO4−质量浓度和pH分别为1 300 mg/L和6.8的条件下,理论所需醋酸钠的初始质量浓度为1.07 g/L,但是微生物不可能将所有电子完全转移给ClO4−,其他的新陈代谢活动也会消耗电子,且溶液中还可能存在其他竞争离子[19],所以实际反应所需的质量浓度远远大于1.07 g/L。在实验中,初始乙酸钠质量浓度分别取0,1.3,3.9,6.5和9.1g/L,对ClO4−的厌氧还原去除率的影响如图2所示。
图2表明:碳源的投加量对ClO4−去除影响显著。乙酸钠投加量为0 g/L时,ClO4−在24 h内的去除率仅为2.2%,基本无还原作用;当初始乙酸钠质量浓度为1.3 g/L时, ClO4−的去除率在16 h内基本稳定在17.0%,表明投加的碳源严重不足;在初始乙酸钠质量浓度为3.9~9.1 g/L时,初始6 h内,由于乙酸钠含量比较充足,ClO4−的还原速率较快,去除率都为35%左右,6 h后,乙酸钠质量浓度为3.9 g/L,体系由于碳源不足,ClO4−去除率随时间增加缓慢;24 h内的去除率为76%,而6.5和9.1 g/L体系中ClO4−的去除率仍保持较快的增长,24 h内去除率都达到97%以上。总体看来,当初始乙酸钠投加量由0 g/L增加到6.5 g/L,高氯酸盐还原速率显著增大,去除率由2.2%迅速提高到98.0%,而当乙酸钠投加量继续增加至9.1 g/L,高氯酸盐还原速率基本不变,去除率为99.6%。因此,乙酸钠的最佳投加量为6.5 g/L,约为ClO4−质量浓度的5倍,且过量投加不会明显促进还原作用。
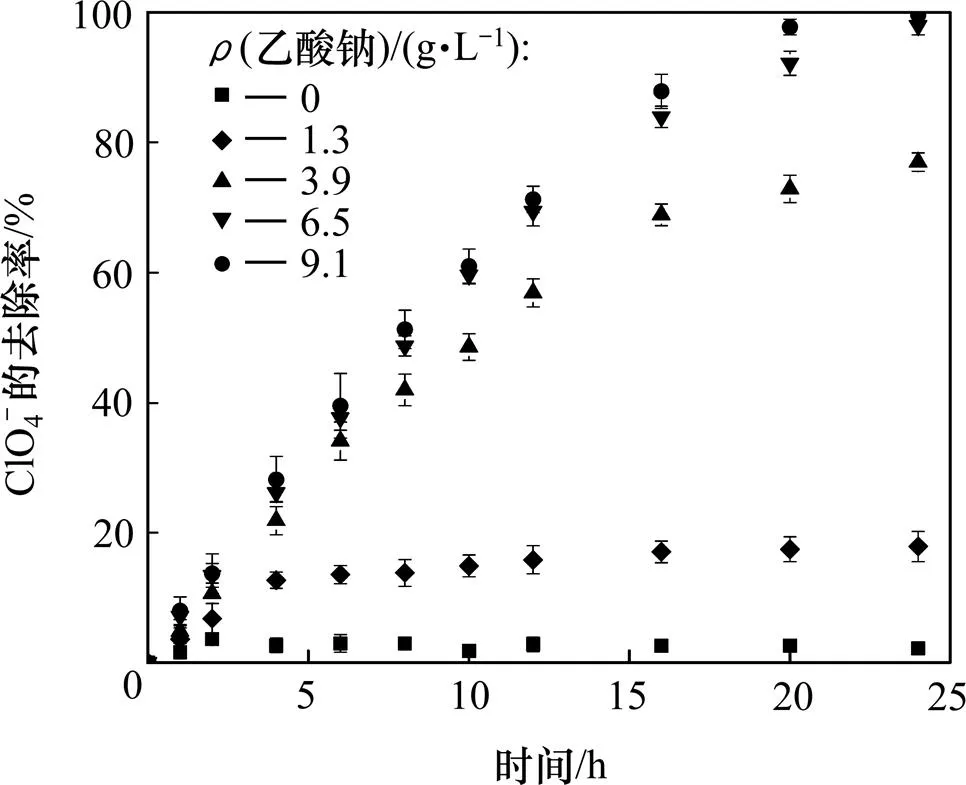
图2 乙酸钠质量浓度对ClO4−去除率的影响
2.3 初始pH和温度对ClO4−还原的影响
pH和温度都是影响微生物代谢活性的重要因素,过高过低都会抑制其新陈代谢速率。据文献报道:在pH为5~9之间,DPRB对ClO4−均有还原作用,并且最适pH在中性范围[20],pH主要通过H+影响ClO4−→ClO3−和ClO3−→ClO2−之间的电子传递,从而影响ClO4−的还原速率[6]。
在初始pH分别为5.55,6.00,6.85,8.00,8.45和9.00下进行实验,得出不同pH对ClO4−平均还原速率的影响,结果如图3所示。图3表明:初始pH对ClO4−去除影响显著。当pH为6~8时,ClO4−还原速率较高,且当pH为6.85时,ClO4−在24 h内的去除率接近100%;当pH偏酸(5~6)或偏碱(8~9)时,ClO4−平均还原速率迅速下降,pH为5.55和8时,ClO4−在24 h内的去除率分别为12.6%与18.0%,而当pH为9时,24 h内基本无ClO4−的还原。因此ClO4−还原的最适pH为6.85左右,系统过酸过碱都不利于微生物的活动,与WU等[12]发现最适pH=8和XU等[6]发现的最适pH=7.5~8.0的结果基本一致。
温度主要是通过改变微生物细胞的生长和还原反应中酶的活性,进而影响高氯酸盐的还原。据报道,温度在10~40℃范围内,DPRB对ClO4−均有还原作用,且最适温度在28~37 ℃之间[20]。在温度为15,20,25,30,35和40 ℃下进行温度对生物还原性能实验,得出温度对ClO4−去除效果的影响,结果如图4所示。

图3 初始pH对高氯酸盐还原速率的影响
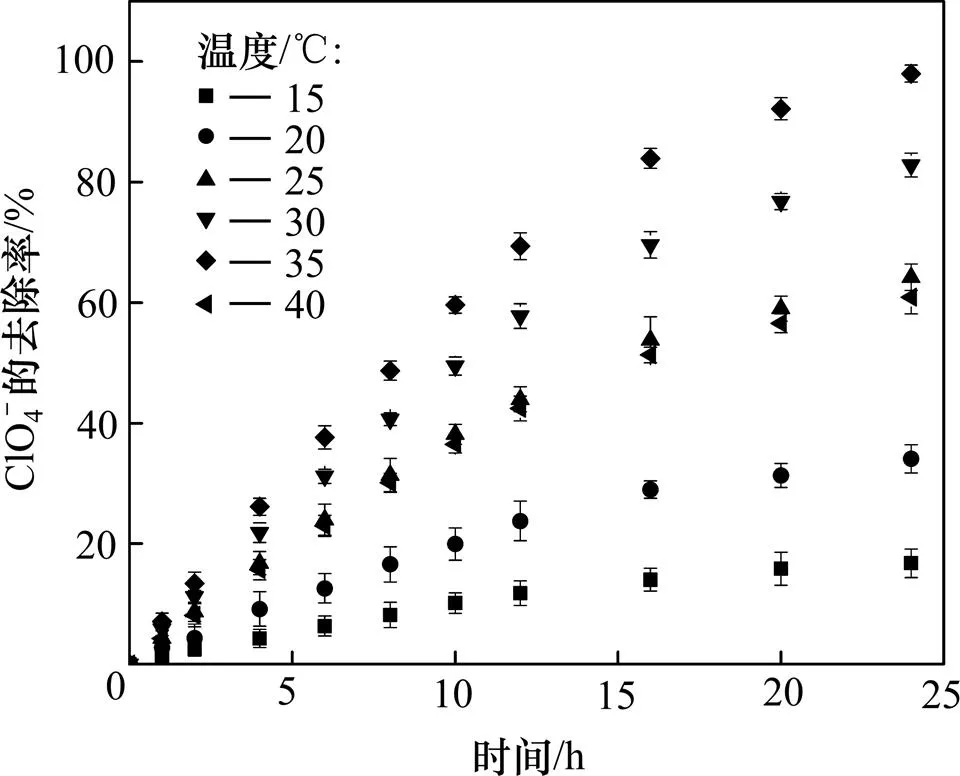
图4 温度对高氯酸盐还原的影响
图4表明:随着温度的升高,高氯酸盐的还原速率和去除率先升高再降低。15 ℃和20 ℃时,ClO4−的还原速率较慢,在24 h内的去除率分别为16.7%和34.1%;25 ℃和30 ℃时,ClO4−的还原速率加快,在24 h内的去除率分别为64.2%和82.8%;35℃时,ClO4−还原速率达到峰值,在24 h内的去除率为98.0%;当温度继续升高到40 ℃时,ClO4−的还原速率有所下降,在24 h内的去除率为60.9%。由此得出DPRB的最适温度为35 ℃,过高或过低都会影响ClO4−的还原速率,此结果与BARDIYA等[20]报道的最适温度在28~37 ℃一致。
2.4 共存离子NO3−和SO42−对ClO4−还原的影响
水中常见阴离子SO42−和NO3−可作为微生物的电子受体参与新陈代谢,是ClO4−还原的竞争离子。理论上,依据各个半反应及其标准电极电势(式(6)~(8))知,θ(ClO4–/ClO3−) 与Eθ(NO3–/HNO2) 比较接近,当NO3−和ClO4−共存时,NO3−是ClO4−还原的主要竞争离子,低质量浓度的NO3−离子也会产生较明显的抑制作用;θ(ClO4–/ClO3−) 是θ(SO42−/H2SO3)的7.7倍,相差较大,当SO42−和ClO4−共存时,SO42−对ClO4−还原的影响较小,但是过量的SO42−仍然能够产生较明显的抑制作用。
ClO4−+2H++2e−→ClO3−+H2O,
θ(ClO4–/ClO3−)=1.23 V (5)
NO3−+3H++2e−→HNO2+H2O,
θ(NO3–/HNO2)=0.93 V (6)
SO42−+4H++2e−→H2SO3+H2O,
θ(SO42−/H2SO3)=0.16 V (7)
本实验中,初始SO42−质量浓度分别为0,1.3,2.6,13.0和26.0 g/L,初始NO3−分别为0,0.65,1.3,2.6和3.9 g/L,探究共存离子对ClO4−去除效果的影响,结果如图5所示。由图5(a)可知:当初始SO42−质量浓度在0~2.6 g/L之间时,ClO4−在12 h内的去除率都在60%以上,24 h内都达到82%以上,保持了较高的去除速率;当初始SO42−质量浓度继续升高到13.0 g/L时,ClO4−在12 h内的去除率达到35.6%,还原速率略有下降,12 h以后,ClO4−的还原速率明显降低,在24 h内的总去除率仅为44.7%,这是由于随着ClO4−的消耗,SO42−质量浓度相对过量,SO42−成为优势离子与ClO4−竞争电子;当初始SO42−质量浓度增加到26.0 g/L,ClO4−在24 h内的去除率为仅为4.5%,ClO4−的还原活动几乎不发生,高质量浓度的SO42−代替ClO4−成为主要的电子受体。总之,当SO42−和ClO4−共存时,SO42−对ClO4−还原的影响较小,只有SO42−质量浓度远大于ClO4−时,才能对ClO4−的还原产生明显的抑制作用。
由图5(b)可知:NO3−对ClO4−还原速率的影响显著。当NO3−质量浓度为0.65 g/L时,ClO4−在10 h内的去除率为7.9%,还原速率缓慢,基本无ClO4−的还原活动,以10 h为折点,ClO4−的还原速率开始增大,到24 h时ClO4−的去除率为51.6%;而NO3−质量浓度为0 g/L NO3−的对照体系,ClO4−在10 h内的去除率已达60%,出现这种现象的原因可能是前10 h内,优先还原NO3−导致其质量浓度降低,在10 h时,NO3−基本被还原完,ClO4−重新成为主要的电子受体,ClO4−的还原活动重新开始;当NO3−质量浓度为1.3和2.6 g/L时,前12 h,ClO4−的去除率分别为4.9%和3.8%,ClO4−的还原基本处于停滞状态,之后以12 h为折点,ClO4−的还原速率开始增大,到24 h时ClO4−的去除率分别为35.9%和29.7%,整体的还原速率仍然较低;当NO3−质量浓度为3.90 g/L时,经过24 h,ClO4−的去除率为3.5%,还原速率很小,ClO4−的还原活动停止。综合来看,初始NO3−质量浓度为0,0.65,1.30,2.60和3.90 g/L的体系中,ClO4−开始发生还原作用的时间分别为0,10,12,12和24 h,初始NO3−质量浓度越高,ClO4−开始发生还原所需的时间越长,说明了NO3−比ClO4−更容易获得电子,微生物优先还原NO3−,再还原ClO4−,这与HERMAN等[21]和何芳等[3]的研究结果一致。

(a) 共存离子SO42−对高氯酸盐还原的影响;(b) 共存离子NO3−对高氯酸盐还原的影响
2.5 高氯酸盐还原菌群的鉴定
通过对驯化污泥的高通量测序分析,得出其中的菌群的分布情况,如图6所示。由图6可知:sp.是驯化污泥中的优势菌群,占总菌的69.32%。sp.是一种常见的ClO4−还原微生物种属[16],能够快速高效的还原高质量浓度高氯酸盐。

图6 富集培养基中的菌群分布
3 结论
1) 在高质量浓度ClO4−条件下驯化得到的DPRB能够快速高效地还原废水中高质量浓度ClO4−(50~ 1 500 mg/L),通过动力学分析得到ClO4−的最大比去除速率max和半饱和常数s远高于之前研究中的结果。因此,本研究得到的高氯酸盐还原菌群具有处理高质量浓度高氯酸盐的潜力。
2) 碳源质量浓度对ClO4−去除影响显著,在初始ClO4−质量浓度为1 300 mg/L时,碳源的最佳投加量为6.5 g/L,且过量投加不会明显促进还原作用,得出碳源的投加量约为ClO4−离子质量浓度的5倍时,ClO4−离子的还原速率最大。
3) 驯化得到的高氯酸盐还原菌对温度、pH较敏感,最适温度和pH分别为35℃和6.85。常见共存离子SO42−和NO3−都会不同程度的抑制高氯酸盐的还原,且NO3−对ClO4−的影响远高于SO42−对ClO4−的 影响。
4)sp.是驯化污泥中的优势菌群,占总菌的69.32%。
参考文献:
[1] SRINIVASAN R, SORIAL G A. Treatment of perchlorate in drinking water: a critical review[J]. Separation and Purification Technology, 2009, 69(1): 7−21.
[2] 王诤, 付学起. 饮用水中的高氯酸盐[J]. 净水技术, 2001, 20(4): 3−4. WANG Zheng, FU Xueqi. Perchlorate in the drinking water[J]. Water Purification Technology, 2001, 20(4): 3−4.
[3] 何芳, 李富生, 周海红, 等. 以改性聚酯颗粒为碳源去除饮用水中高氯酸盐[J]. 中南大学学报(自然科学版), 2014, 45(10): 4452−4457. HE Fang, LI Fusheng, ZHOU Haihong, et al. Removal of perchlorate from drinking water using modified polyester particles as carbon source[J]. Journal of Central South University (Science and Technology), 2014, 45(10): 4452−4457.
[4] SUSARLA S, COLLETTE T W, GARRISON A W, et al. Perchlorate identification in fertilizers[J]. Environmental Science & Technology, 1999, 33(19): 3469−3472.
[5] WOLFF J. Perchlorate and the thyroid gland[J]. Pharmacological Reviews, 1998, 50(1): 89−106.
[6] XU Xing, GAO Baoyun, JIN Bo, et al. Study of microbial perchlorate reduction: considering of multiple pH, electron acceptors and donors[J]. Journal of Hazardous Materials, 2015, 285(3): 228−235.
[7] DUDLEY M, SALAMONE A, NERENBERG R. Kinetics of a chlorate-accumulating, perchlorate-reducing bacterium[J]. Water Research, 2008, 42(1): 2403−2410.
[8] RIKKEN G B, KROON A G M, Van GINKEL C G. Transformation of (per) chlorate into chloride by a newly isolated bacterium: reduction and dismutation[J]. Applied Microbiology and Biotechnology, 1996, 45(3): 420−426.
[9] 江毅. 生物法还原水中高氯酸盐的研究[D]. 广州: 华南理工大学环境与能源学院, 2013: 26−29. JIANG Yi. Research of perchlorate reduction in water by bacteria[D]. Guangzhou: South University of Technology. School of Environment and Energy, 2013: 26−29.
[10] WANG Chao, LEE L, MENG Xiaoguang. Kinetics of biological perchlorate reduction and pH effect[J]. Journal of Hazardous Materials, 2008, 153(1): 663−669.
[11] DUGAN N R, WILLIAMS D J, MEYER M, et al. The impact of temperature on the performance of anaerobic biological treatment of perchlorate in drinking water[J]. Water research, 2009, 43(7): 1867−1878.
[12] WU Donglei, HE Ping, XU Xinhua, et al. The effect of various reaction parameters on bioremediation of perchlorate- contaminated water[J]. Journal of Hazardous Materials, 2008, 150(2): 419−423.
[13] BATISTA J R, MCGARVEY F X, VIEIRA A R. The removal of perchlorate from waters using ion-exchange resins, Perchlorate in the Environment [M]. New York: Springer US, 2000: 135−145.
[14] HATZINGER P B. Perchlorate biodegradation for water treatment[J]. Environmental Science & Technology, 2005, 39(11): 239A−247A.
[15] NERENBERG R, KAWAG Y, RITTMANN B E. Microbial ecology of a perchlorate-reducing, hydrogen-based membrane biofilm reactor[J]. Water research, 2008, 42(4): 1151−1159.
[16] NOR S J, LEE S H, CHO K S, et al. Microbial treatment of high-strength perchlorate wastewater[J]. Bioresource Technology, 2011, 102(2): 835−841.
[17] 王蕊, 刘菲, 陈鸿汉, 等. 电子供体对地下水中高氯酸盐生物去除的影响研究[J]. 环境科学学报, 2013, 33(11): 3060−3067. WANG Rui, LIU Fei, CHEN Honghan, et al. Electron donors for the biological removel of perchlorate in groundwater[J]. Acta Scientiae Circumstantiae, 2013, 33(11): 3060−3067.
[18] COATES J D, JACKSON W A. Principles of perchlorate treatment., In situ Bioremediation of perchlorate in groundwater[M]. New York: Springer New York, 2009: 29−53.
[19] 钱慧静, 奚胜兰, 何平, 等. 生物法还原高氯酸盐及其优化研究[J]. 环境科学, 2009, 30(5): 1402−1407. QIAN Huijing, XI Shenglan, HE Ping, et al. Biological reduction of perchlorate and optimization[J]. Environmental Science, 2009, 30(5): 1402−1407.
[20] BARDIYA N, BAE J H. Dissimilatory perchlorate reduction: a review[J]. Microbiological Research, 2011, 166(4): 237−254.
[21] HERMAN D C, FRANKENBERGER W T. Microbial-mediated reduction of perchlorate in groundwater[J]. Journal of Environmental Quality, 1998, 27(4): 750−754.
(编辑 陈爱华)
Kinetics of biological high-concentration perchlorate reduction and optimization of reaction conditions
WU Min, WANG Shuaifeng, GAO Naiyun, ZHU Yanping, LI Shuo, ZHU Sirui
(State Key Laboratory of Pollution Control and Resource Reuse, Tongji University, Shanghai 200092, China)
To treat wastewater containing high-mass concentration perchlorate (ClO4−), heterotrophic and perchlorate-reducing bacteria were obtained by acclimating anaerobic activated sludge grown on high-mass concentration perchlorate (1 500 mg/L) feed medium. Batch experiments were conducted to investigate the kinetics of biological high- mass concentration perchlorate reduction and the effect of initial perchlorate mass concentrations (50−1 500 mg/L), acetate mass concentrations (1.69~11.83 g/L), pH (5.55−9.00), temperature (15−40℃), common coexistence SO42−(0−26.0 g/L) and NO3−(0−3.9 g/L) on perchlorate reduction. The results show that the maximum specific perchlorate reduction rate (max) and half saturation constant (s) are 0.89 (mg·mg)/h and 141.6 mg/L, respectively, which indicates that the cultures enriched are effective at treating high-strength perchlorate wastewater. Under the selected conditions, namely 6.5 g/L acetate (the mass concentration ratio of CH3COONa to ClO4−is 6), initial pH 6.85 and 35℃, 1 300 mg/L perchlorate can be rapidly reduced to non-detectable levels within 24 h. Both common coexistence SO42−and NO3−can inhibit the rate and extent of perchlorate reduction, especially NO3−. The dominant perchlorate reducing bacteria in the consortium issp. (69.32%).
perchlorate; high mass concentration; perchlorate reducing bacteria; kinetics; optimization
10.11817/j.issn.1672-7207.2016.11.045
X52
A
1672−7207(2016)11−3958−07
2016−01−09;
2016−04−15
国家科技重大专项资助(2012ZX07403-001); 国家自然科学基金资助项目(51178321); 住房和城乡建设部研究开发项目(2009-K7-4) (Project(2012ZX07403-001) supported by National Major Science and Technology Project of China; Project(51178321) supported by National Natural Science Foundation of China; Project(2009-K7-4) supported bythe Research and Development Project of Ministry of Housing and Urban-Rural Development of China)
吴敏,博士,副教授,从事水污染控制研究和资源化研究;E-mail: minw@tongji.edu.cn
猜你喜欢
能源与环境(2022年5期)2023-01-10
中国资源综合利用(2022年9期)2022-10-13
氯碱工业(2022年1期)2022-07-02
现代矿业(2022年3期)2022-04-09
现代牧业(2020年4期)2020-12-31
制造技术与机床(2019年9期)2019-09-10
中国氯碱(2017年4期)2017-05-04
军事文摘·科学少年(2016年10期)2016-12-08
军事文摘(2016年20期)2016-11-07
饮食科学(2015年7期)2015-11-23
