
多靶点药物治疗前列腺癌研究进展
2016-12-22 02:39吴萌谢永丽岑山周金明
中国医药生物技术 2016年6期
吴萌,谢永丽,岑山,周金明
多靶点药物治疗前列腺癌研究进展
吴萌,谢永丽,岑山,周金明
前列腺癌是男性群体中常见癌症,在所有癌症种类中,其致死率位列第六[1]。治疗早期前列腺癌常用外科手术摘除和放射疗法然后辅以药物治疗,但是对于去势性抵抗前列腺癌(castration resistant prostate cancer,CRPC),药物治疗仍然是首选策略。根据与前列腺癌有关的生物信号通路及靶标来开发抗前列腺癌药物是目前该类药物研发的常用方法。随着技术的进步,越来越多的研究表明,药物的多靶点效应是药物发挥药效的不可或缺的因素[2](图 1)。多靶点药物的研发近年来日益受到研究者的重视,尤其是针对癌症、神经退行性疾病以及心血管疾病等发病机制复杂的疾病,多靶点药物拥有独特优势[3]。本文回顾了最近几年多靶点药物在治疗前列腺癌领域的研究及应用进展,以期能帮助研究者进一步认识该领域的相关情况。
1 多靶点药物的发展
药物的多靶点效应,即单个药物作用于一个或多个通路的多个生物靶标,曾被认为是药物产生副作用或毒性的主要原因。随着技术的进步,越来越多的研究表明,药物的多靶点效应是其发挥药效的不可或缺的因素。网络生物学认为,对于复杂的生物系统,即使是单靶点药物已经完全改变了它们所作用靶标的功能,这些药物可能依然很难达到人们所预期的效果。因为生物系统往往拥有该靶点的“备份”系统,并且很多细胞网络具有稳健的特征,即使是网络中某个节点发生巨大改变,网络整体功能也不会发生较大变化。此外,很多疾病是由多种致病因素所引起的,例如阿尔茨海默病、帕金森综合征等神经退行性疾病以及各种癌症[2]。因此,对于这些发病机制复杂的疾病,仅仅阻断单个靶点,药物很难发挥作用。
为了同时作用于多个靶点,主要有两种选择:使用多靶点药物或多个单靶点药物的联合用药。到目前为止,联合用药已经在几种疾病的治疗中取得了成功,例如治疗 HIV 的鸡尾酒疗法,Hsp90 抑制剂与其他药物联合使用治疗实体肿瘤与白血病等。相比于单靶点药物的联合用药,多靶点药物具有如下优势:多靶点药物是单分子药物,其体内药代动力学更好预测,因此,可能具有良好的药代动力学特征和安全性。此外,多靶点药物可以在机体各组织中同时达到动态平衡,有利于药物同时作用于各个靶点,更好达到协同增效的目的。并且使用多靶点药物可以避免产生联合用药中可能出现的药物—药物相互作用[4]。因此,多靶点药物的发展前景值得期待。
2 治疗前列腺癌的相关通路与靶点
雄激素信号通路在前列腺癌形成及发展过程中发挥着重要作用[5]。雄激素的体内分泌主要受下丘脑—垂体—性腺通路和下丘脑—垂体—肾上腺通路调控,在两条通路调控下,在肾上腺和睾丸中,由 17α-羟化酶/C17,20-裂合酶(class steroid 17α-hydroxylase/C17,20-lyase,CYP17)以及其他酶作用下生成睾酮和脱氢表雄酮(dehydroepiandrosterone,DHEA),DHEA 及其硫酸盐在外围组织中经羟化类固醇脱氢酶(3βHSD、17βHSD 等)连续催化下生成睾酮。随后,5α-还原酶(-reductase)将产生的睾酮催化还原成二氢睾酮(dihydrotestosterone,DHT)[6-7]。DHT 与雄激素受体(androgen receptor,AR)结合,诱导 AR N-C 相互作用并形成二聚体,AR 二聚后转入细胞核与靶基因结合,调控一系列转录反应,促进前列腺癌细胞生长增殖(图 2)。

图 1 单靶点药物与多靶点药物

图 2 治疗前列腺癌的主要通路及靶点
磷脂酰肌醇-3-激酶(phosphatidylinositol-3-hydroxy kinase,PI3K)信号通路(图 2)是调控许多细胞生理机能的关键信号转导通路,主要包括 PI3K/AKT/mTOR 几个关键节点[7],该通路在前列腺癌细胞的生长、生存、黏附和转移中均发挥着重要作用,尤其表现在癌症的演进、转移扩散以及辐射抵抗方面。在外部因子(如生长因子、性激素)的刺激下,PI3K 集中分布于细胞膜脂质基质磷脂酰肌醇二磷酸(PIP2)处,并催化 PIP2 转化为磷脂酰肌醇三磷酸(PIP3),PIP3 使 PI3K 下游效应器 AKT/PKB 水平增加,蛋白激酶 B(protein kinase B,AKT)通过激活哺乳动物雷帕霉素靶蛋白(mTOR),进而引起下游一系列的转录反应,促进癌细胞的蛋白质合成及生长增殖[8-9]。
细胞凋亡阻滞效应是晚期癌症产生抗性的重要原因,在前列腺癌患者中,有许多蛋白与此效应相关,其中包括细胞凋亡调控因子 Bcl-2、丛生蛋白(clusterin)和 AKT1[10]。除此之外,细胞凋亡抑制蛋白(inhibitor of apoptosis protein,IAP)家族也发挥着重要作用,其中包括cIAP1、cIAP2、XIAP 以及生存蛋白和 BIRC6 等,这些 IAP 家族成员在前列腺癌患者中表达水平均有所增加[11]。下面以 Bcl-2 蛋白家族为例介绍下该类蛋白抑制细胞凋亡的机制。Bcl-2 所在的蛋白家族包括两类蛋白质:一类是抗凋亡蛋白(Bcl-2、Bcl-xL、Bcl-w、Mcl-1 和 A1),另一类是促凋亡蛋白(Bax、Bak、Bad、Bid 和 Bcl-xS)。在细胞凋亡时,Bcl-2 家族中的促凋亡蛋白成员发生蛋白质的加工修饰,易位到线粒体的外膜上,引起细胞色素 c、凋亡诱导因子等其他促凋亡因子的释放,导致细胞凋亡(图 2);而该家族的抗凋亡蛋白成员则平时被隔离在线粒体等细胞器内,可以抑制细胞色素 c 和凋亡诱导因子等促凋亡因子的释放,具有抑制细胞凋亡的功能[12]。
3 基于信号通路的多靶点抗前列腺癌药物
3.1 雄激素信号通路抑制剂
阻断雄激素信号通路是治疗前列腺癌的重要策略之一。美国 FDA 2013 年批准上市的抗前列腺癌药物阿比特龙(abiraterone)是第一个上市的阻断雄激素合成的多靶点抗前列腺癌药物。阿比特龙(图 3)可以通过作用于 CYP17、3βHSD来阻断雄激素的生成,此外它还是 AR 拮抗剂,通过协同作用于多个靶点,阿比特龙明显延长了患者的生存期[13]。阿比特龙的成功上市鼓舞了研究人员对于该类药物的研发信心。Li 等[14]研究了阿比特龙体内代谢产物 D4A(图 3),发现与阿比特龙相比,D4A 具有更好的体外与体内抗前列腺癌活性。D4A 的作用靶点与阿比特龙基本相同,主要包括 CYP17、3βHSD 和 5α-还原酶,通过与 AR 拮抗剂恩杂鲁胺作对照试验,结果证明 D4A 也具有拮抗 AR 的作用,直接利用 D4A 治疗前列腺癌可能会收到更佳疗效。
Galeterone(TOK-001 或 VN/124-1)(图 3)同样是类固醇类药物,其化学结构与阿比特龙相似。Galeterone 的体外活性实验表明,它阻断 CYP17 催化活性的效果要优于阿比特龙(IC50为 300 nmol/L)。此外,galeterone 还能竞争性地与 AR 结合从而抑制 AR 的功能。小鼠异种移植肿瘤实验显示,galeterone 能明显抑制 LAPC4 前列腺肿瘤组织的形成与生长,临床实验表明其具有良好剂量耐受性,用药后 PSA 水平下降超过 50% 的患者占了 75%[15-16]。在长期缺乏雄激素的晚期前列腺癌患者中,AR 往往会发生突变以适应低雄激素环境。此时,AR 拮抗剂或者人体内源性皮质醇等类固醇化合物都有可能成为 AR 激动剂。

阿比特龙 D4A Galeterone 化合物 6
化合物 10(R=OMe) BEZ235 PI103 BGT226
化合物 11(R=OH)
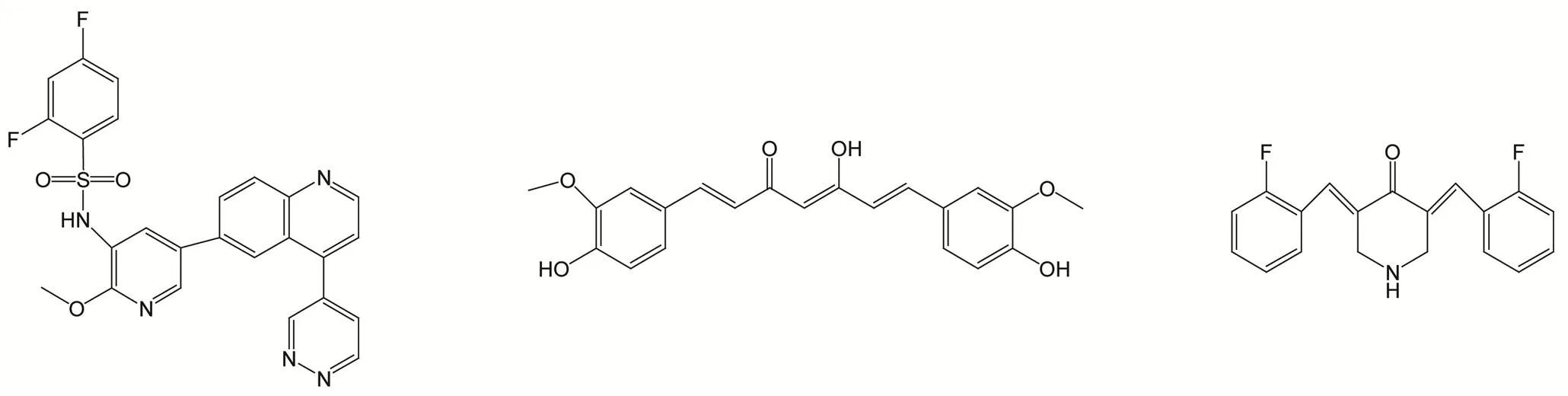
GSK2126458 Curcumin EF24
化合物 3b
图 3 治疗前列腺癌的多靶点化合物结构
此外,雄激素的缺乏还会导致醛固酮水平的升高,进而造成一系列心血管并发症[17-18]。CYP11B1 与 CYP11B2 分别可以催化皮质醇与醛固酮的最后生物合成步骤,因此开发同时抑制 CYP17 和 CYP11B1、CYP11B2 的药物,或许将有助于降低上述副作用。Hu 等[17]设计合成了一系列联苄基烯吡啶类化合物,通过活性筛选,发现化合物 6(图 3)具有CYP17/CYP11B1 双抑制活性(IC50为 226 和 287 nmol/L)。Pinto-Bazurco Mendieta 等[18]设计合成了一系列非类固醇化合物,其中化合物 10 和 11(图 3)具有 CYP17/CYP11B2 双抑制活性。通过对这几个化合物的研究,为多靶点雄激素通路抑制剂的研发提供了新思路。
3.2 PI3K 信号通路抑制剂
PI3K 信号通路在晚期前列腺癌发展中具有重要作用,同时阻断该信号通路的不同节点以抑制癌细胞生长增殖,是治疗前列腺癌的策略之一。化合物 BEZ235(图 3)对PI3K 与 mTOR 具有双抑制活性,与它的作用靶点相同的还有化合物 PI103(图 3)。Chang 等[19]利用三组辐射抵抗型的前列腺癌(CaP-RR)细胞系(PC-3RR、DU145RR 和 LNCaPRR)研究发现,BEZ235 和 PI103 与放射疗法合用,可以导致 CaP-RR 细胞系自噬减少、细胞循环阻滞、DNA 损伤修复功能减退、细胞凋亡数目增加等一系列细胞损伤作用,从而提高辐射抵抗前列腺癌细胞对于放射治疗的敏感性。并且与选择性抑制 PI3K 的 BKM120 和选择性抑制 mTOR 的雷帕霉素相比,具有 PI3K 与 mTOR 双抑制活性的 BEZ235 和 PI103,表现出了更高的肿瘤抑制活性。
BGT226(图 3)是一个具有 PI3K 与 mTOR 双抑制活性的咪唑喹啉衍生物,研究发现,BGT226 可以抑制 p110α, β, δ, γ 等 PI3K 酶以及 mTOR 激酶,而对于其他酶则没有明显抑制作用。膜过滤结合分析实验显示,BGT226 与 PI3K 各个亚型的 IC50值均小于 100 nmol/L。细胞实验显示,BGT226 抑制肿瘤细胞增殖的活性在纳摩尔水平。包括前列腺癌在内的多种异种肿瘤移植实验证明,该化合物能抑制体内固体瘤的生长,临床I 期试验结果显示,BGT226 体内药代动力学良好[20]。
GSK2126458(图 3)是一个高活性的 PI3K/mTOR 双抑制剂,它可以选择性地抑制 PI3K 与 mTOR 的多个亚型,包括 p110α、p110β、p110γ、p110δ、mTORC1、mTORC2 等。GSK2126458 可以明显降低磷酸化 AKT 水平,抑制乳腺癌细胞生长,导致细胞有丝分裂 G1 期阻滞[21]。此外,有文献报道 GSK2126458 在治疗固体瘤与淋巴瘤方面也已经进入了临床 I 期实验[22]。Park 等[23]研究发现,将该化合物与 MEK 抑制剂 AZD6244 联合使用,可以同时抑制 RAS/RAF/MEK/ERK 和 PI3K/AKT/mTOR 两条信号通路。体外与体内实验表明,两者合用既可以降低磷酸化 AKT水平,又可以降低磷酸化 EPK 水平,从而激活一系列细胞损伤作用,抑制前列腺癌细胞 DU145 和 PC-3 的生长和增殖。
基于以上作用于雄激素信号通路与 PI3K 信号通路的多靶点药物,我们不难发现,相比于单靶点药物,通过协同抑制机体信号通路的多个节点,多靶点药物可以增强治疗效果。因此,基于信号通路中的不同靶点来理性设计可以同时作用于多个靶点的药物具有广阔应用前景。
4 多靶点反义寡核苷酸药物
反义寡核苷酸是一类新型药物,它通过结合特定的mRNA阻碍 mRNA 编码的靶蛋白生成,进而降低与目标疾病相关的蛋白表达量,以达到治疗疾病的目的[24]。通过设计合成双特异反义寡核苷酸,同时作用于两个 mRNA,可以更好地达到协同治疗效果。
Rubenstein 等[10]设计合成了抑制 Bcl-2 表达的反义寡核苷酸 MR24、MR42 与MR4。结果发现,前列腺癌细胞可以通过各种补偿途径对其产生抗药性。其中增殖抗原蛋白 KI-67 的过表达在各种补偿途径中占有突出地位,并发现研发同时作用于 Bcl-2 与 KI-67 的双特异反义寡核苷酸,能更有效地促进前列腺癌细胞凋亡。
Bcl-2 与 Bcl-xL 作为抗凋亡蛋白,在抑制肿瘤细胞凋亡方面发挥着重要作用。Yamanaka 等[25]设计了能够抑制Bcl-2 与 Bcl-xL 表达的双特异反义寡核苷酸,实验表明,该双特异反义寡核苷酸能够降低 Bcl-2 与 Bcl-xL 基因表达的 mRNA 与蛋白水平,进而促进细胞凋亡、抑制肿瘤细胞增长。将该双特异反义寡核苷酸与其他传统治疗前列腺癌方法联合使用,增加了前列腺癌LNCaP 细胞对于其他疗法的敏感性。值得注意的是,在Bcl-2 与 Bcl-xL 得到抑制的同时,抗凋亡蛋白丛生蛋白表达水平显著增加,因此,若能抑制该蛋白的表达量对于促进肿瘤细胞凋亡有重要意义。Custirsen(OGX-011)是一个特异性作用于丛生蛋白 mRNA 的反义寡核苷酸[26],若以 custirsen 为基础,通过理性设计方法,设计出可以同时作用于丛生蛋白 mRNA 以及 Bcl-2 或 Bcl-xL mRNA 的双特异反义寡核苷酸,其结果值得期待。
从调控细胞凋亡抑制蛋白(IAP)家族成员出发,Luk等[11]设计合成了一系列双特异反义寡核苷酸。结果发现,6w2 能够抑制 BIRC6 和 cIAP1 的表达量,而 6w5 能够抑制 BIRC6 和生存素的表达量。与仅能抑制单个蛋白表达的对照组相比,6w2 和 6w5 明显阻滞了 PC-3 与 C4-2 细胞增殖,促进了前列腺癌细胞的凋亡。PC-3 肿瘤细胞异种移植实验表明,这两个双特异反义寡核苷酸可以导致肿瘤组织坏死率显著增加。
5 多靶点中药来源化合物
在药物发现过程中,自然产物及其结构衍生物发挥了重要作用,中药在中国有广泛的应用,并且在西方国家也被作为先导化合物的重要来源之一[27]。许多中药化合物在体内具有多靶点效应,因此针对中药多靶点化合物的研究逐渐被重视。
Curcumin(图 3)是姜黄的主要成分之一。研究发现,curcumin 可以抑制多种肿瘤细胞和组织的生长与转移扩散。Liu 等[28]利用 PC-3 前列腺癌细胞的研究结果表明,curcumin 通过阻断NF-κB信号通路和 AP-1 信号通路的功能,导致细胞循环阻滞、抑制细胞生长和促进细胞凋亡。此外,Sundram 等[29]报道了curcumin通过激动蛋白激酶 D1(PKD1)来抑制细胞核内的β-连环蛋白信号通路,从而抑制原癌基因的表达,降低癌细胞的运动能力。体内实验显示,curcumin 明显抑制了前列腺癌细胞瘤的生长。EF24(图 3)是curcumin的结构类似物,它同样可以抑制NF-κB信号通路。此外,EF24还可以通过作用于 miRNA-21,抑制其表达水平,从而促进 miRNA-21 调控的靶基因 PTEN 和 PDCD4 的表达。EF24 表现出了明显的前列腺癌细胞抑制活性,促进癌细胞凋亡。前列腺癌细胞 DU145 异种移植实验表明,EF24 具有良好的体内抑瘤活性,它可以明显降低肿瘤组织的生长速度[30]。
β-紫罗兰酮是水果、蔬菜和谷物的化学成分之一,研究发现它有体外抗肿瘤活性,Zhou 等[31]将 β-紫罗兰酮与curcumin 合并成单体化合物,并以此为母核设计合成了一系列中药来源化合物的衍生物。研究发现,其中的一些化合物不仅有拮抗 AR 的活性,化合物 SC97 和 SC245 还有抑制 NF-κB 激活的作用。Liu 等[32]进一步对这两个化合物进行结构优化,得到了活性更高的化合物 3b(图 3)。通过拮抗 AR 与抑制 NF-κB 的激活,化合物 3b 可以明显降低 PSA 表达水平,导致 LNCaP 细胞凋亡,并且防止多种前列腺癌细胞的增殖扩散。
白细胞介素-6(IL-6)与 AR 的激活有关,是治疗前列腺癌的潜在靶点之一。有研究报道,穿心莲内酯可以在 mRNA 与蛋白水平抑制 IL-6 的表达。进一步的研究发现,穿心莲内酯也可以直接抑制 AR 信号通路,它可以在 mRNA 与蛋白水平降低 AR 的表达,阻断 Hsp-90 与 AR 的结合。通过作用于以上靶点,穿心莲内酯可以促进 AR 降解,抑制前列腺癌细胞生长,导致细胞凋亡增加,体内实验显示,它可以抑制前列腺癌细胞瘤的生长[33-34]。
6 小结
传统治疗手段对于晚期前列腺癌的治疗效果不佳。随着人们对于前列腺癌发病机制认识的不断深入,通过设计作用于不同信号通路与生物靶标的多靶点药物,来协同抑制前列腺癌的发展,是当前研究方向之一。传统中药来源化合物与反义寡核苷酸药物可以作为多靶点药物的重要来源,通过进一步优化有可能得到活性较好的药物。根据雄激素信号通路与其他信号通路,理性设计多靶点药物也取得了重要进展。在对前列腺癌相关靶点研究的基础上,综合利用网络生物学、结构生物学、药物化学以及计算机辅助药物设计等技术,开发能够应用于临床的治疗前列腺癌的多靶点药物,将会对抗前列腺癌药物的研发带来新希望。
[1] Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med, 2015, 373(18):1697- 1708.
[2] Zhou J, Li Q, Wu M, et al. Progress in the rational design for polypharmacology drug. Curr Pharm Des, 2016, 22(21):3182-3189.
[3] Guo YS, Guo ZR. Design of multiple targeted drugs. Acta Pharm Sinica, 2009, 44(3):276-281. (in Chinese)
郭彦伸, 郭宗儒. 多靶点药物分子设计. 药学学报, 2009, 44(3): 276-281.
[4] Medina-Franco JL, Giulianotti MA, Welmaker GS, et al. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today, 2013, 18(9-10):495-501.
[5] Dong Y, Zhang H, Gao AC, et al. Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol Cancer Ther, 2005, 4(7):1047-1055.
[6] Franchimont P. Regulation of gonadal androgen secretion. Horm Res, 1983, 18(1-3):7-17.
[7] Rainey WE, Nakamura Y. Regulation of the adrenal androgen biosynthesis. J Steroid Biochem Mol Biol, 2008, 108(3-5):281-286.
[8] Mazzoletti M, Bortolin F, Brunelli L, et al. Combination of PI3K/mTOR inhibitors: antitumor activity and molecular correlates. Cancer Res, 2011, 71(13):4573-4584.
[9] Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol, 2010, 28(6):1075-1083.
[10] Rubenstein M, Hollowell CM, Guinan P. Suppression of BCL2 by antisense oligonucleotides and compensation by non-targeted genes may enhance tumor proliferation. In vivo, 2015, 29(6):687-694.
[11] Luk SU, Xue H, Cheng H, et al. The BIRC6 gene as a novel target for therapy of prostate cancer: dual targeting of inhibitors of apoptosis. Oncotarget, 2014, 5(16):6896-6908.
[12] Fu YF, Fan TJ. Bcl-2 family proteins and apoptosis. Acta Biochimica Et Biophysica Sinica, 2002, 34(4):389-394. (in Chinese)
付永锋, 樊廷俊. Bcl-2家族蛋白与细胞凋亡. 生物化学与生物物理学报, 2002, 34(4):389-394.
[13] Yin L, Hu Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol, 2014, 11(1):32-42.
[14] Li Z, Bishop AC, Alyamani M, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature, 2015, 523(7560):347-351.
[15] Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem, 2015, 58(5):2077-2087.
[16] Purushottamachar P, Godbole AM, Gediya LK, et al. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J Med Chem, 2013, 56(12):4880-4898.
[17] Hu Q, Jagusch C, Hille UE, et al. Replacement of imidazolyl by pyridyl in biphenylmethylenes results in selective CYP17 and dual CYP17/CYP11B1 inhibitors for the treatment of prostate cancer.J Med Chem, 2010, 53(15):5749-5758.
[18] Pinto-Bazurco Mendieta MA, Hu Q, Engel M, et al. Highly potent and selective nonsteroidal dual inhibitors of CYP17/CYP11B2 for the treatment of prostate cancer to reduce risks of cardiovascular diseases. J Med Chem, 2013, 56(15):6101-6107.
[19] Chang L, Graham PH, Hao J, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis, 2014, 5:e1437.
[20] Markman B, Tabernero J, Krop I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol, 2012, 23(9):2399- 2408.
[21] Leung E, Kim JE, Rewcastle GW, et al. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther, 2011, 11(11): 938-946.
[22] Liu T, Sun Q, Li Q, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther, 2015, 14(2):429-439.
[23] Park H, Kim Y, Sul JW, et al. Synergistic anticancer efficacy of MEK inhibition and dual PI3K/mTOR inhibition in castration-resistant prostate cancer. Prostate, 2015, 75(15):1747-1759.
[24] Hnik P, Boyer DS, Grillone LR, et al. Antisense oligonucleotide therapy in diabetic retinopathy. J Diabetes Sci Technol, 2009, 3(4): 924-930.
[25] Yamanaka K, Rocchi P, Miyake H, et al. Induction of apoptosis and enhancement of chemosensitivity in human prostate cancer LNCaP cells using bispecific antisense oligonucleotide targeting Bcl-2 and Bcl-xL genes. BJU Int, 2006, 97(6):1300-1308.
[26] Chi KN, Zoubeidi A, Gleave ME. Custirsen (OGX-011): a second-generation antisense inhibitor of clusterin for the treatment of cancer. Expert Opin Investig Drugs, 2008, 17(12):1955-1962.
[27] Li-Weber M. Targeting apoptosis pathways in cancer by Chinese medicine. Cancer Lett, 2013, 332(2):304-312.
[28] Liu S, Wang Z, Hu Z, et al. Anti-tumor activity of curcumin against androgen-independent prostate cancer cells via inhibition of NF-κB and AP-1 pathway in vitro. J Huazhong Univ Sci Technolog Med Sci, 2011, 31(4):530-534.
[29] Sundram V, Chauhan SC, Ebeling M, et al. Curcumin attenuates β-catenin signaling in prostate cancer cells through activation of protein kinase D1. PLoS One, 2012, 7(4):e35368.
[30] Yang CH, Yue J, Sims M, et al. The Curcumin analog EF24 targets NF-kB and miRNA-21, and has potent anticancer activity in vitro and in vivo. PLoS One, 2013, 8(8):e71130.
[31] Zhou J, Geng G, Shi Q, et al. Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. J Med Chem, 2009, 52(17):5546-5550.
[32] Liu W, Zhou J, Geng G, et al. Synthesis and in vitro characterization of ionone-based compounds as dual inhibitors of the androgen receptor and NF-κB. Invest New Drugs, 2014, 32(2):227-234.
[33] Chun J, Tummala R, Nadiminty N, et al. Andrographolide, an herbalmedicine, inhibits interleukin-6 expression and suppresses prostate cancer cell growth. Genes Cancer, 2010, 1(8):868-876.
[34] Liu C, Nadiminty N, Tummala R, et al. Andrographolide targets androgen receptor pathway in castration-resistant prostate cancer. Genes Cancer, 2011, 2(2):151-159.
国家自然科学基金(81001402、81311120299、81672559)
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所免疫生物学室
周金明,Email:zhoujinming@imb.pumc.edu.cn;岑山,Email:shancen@hotmail.com
2016-08-04
10.3969/j.issn.1673-713X.2016.06.008
猜你喜欢
中老年保健(2021年3期)2021-12-03
小学生学习指导·低年级(2021年6期)2021-09-10
考试与评价·七年级版(2021年4期)2021-08-14
中国生殖健康(2020年7期)2020-12-10
天津医科大学学报(2019年3期)2019-08-13
中国生殖健康(2019年7期)2019-01-06
小学阅读指南·低年级版(2018年5期)2018-11-02
中国男科学杂志(2016年5期)2016-12-01
中国男科学杂志(2016年5期)2016-12-01
医学研究杂志(2015年7期)2015-06-22
