
银黄口服液含量测定方法的改进试验
2016-12-21 10:40周艳飞王亚芳李应超王海军
中国兽医杂志 2016年10期
周艳飞 , 李 浛 , 王亚芳,李应超 , 魏 镭 , 王海军
(1.北京市兽药监察所 , 北京大兴102629;2.北京生泰尔生物科技有限公司 ,北京大兴102206 ; 3.北京市中兽药工程技术研究中心 , 北京大兴102206)
银黄口服液含量测定方法的改进试验
周艳飞1, 李 浛1, 王亚芳1,李应超1, 魏 镭1, 王海军2,3
(1.北京市兽药监察所 , 北京大兴102629;2.北京生泰尔生物科技有限公司 ,北京大兴102206 ; 3.北京市中兽药工程技术研究中心 , 北京大兴102206)
为了建立同时测定银黄口服液中绿原酸和黄芩苷的高效液相色谱方法,采用Discovery C18柱(4.6×150 mm,5 μm),以乙腈-0.4%磷酸溶液为流动相进行梯度洗脱,流速为1.0 mL/min,进样量为10 μL,检测波长327 nm。结果目标峰分离度良好,绿原酸和黄芩苷的质量浓度与峰面积分别在1.00~20.05 μg/mL(R2=0.999 9)、10.07~201.55 μg/mL(R2=0.999 9)呈良好线性,平均加样回收率分别为99.90%和100.73%,标准偏差(RSD)均小于1.0%。表明此方法重复性好,专属性强,可准确、快速测定银黄口服液中的绿原酸和黄芩苷含量。
银黄口服液 ; 高效液相色谱法 ; 黄芩苷 ; 绿原酸
银黄口服液是由黄芩与金银花提取加工而成,具有抗菌、清热解毒、消炎、退热等作用,临床上广泛应用于上呼吸道感染、急慢性扁桃体炎等症的治疗,疗效确切[1]。银黄口服液最早被列入新药转正标准第2册(1993年)中,自2000年以来被历版中国药典收载。中国药典(2010年版)现行质量标准中,分别进行金银花和黄芩苷的含量测定,检测时间相对长[2]。
目前,已有相关研究对银黄口服液中绿原酸和黄芩苷进行了同时含量测定,方法报道不一[3-6]。本试验根据国家兽药典委员会2015版兽药典标准的要求,对银黄口服液中绿原酸和黄芩苷同时测量方法进行了系统的研究,为新版兽药典中银黄口服液质量标准变更提供试验和数据支持。
1 材料
2695高效液相色谱仪,2487紫外检测器(Waters 公司);1100高效液相色谱仪,VWD紫外检测器(Agilent公司);e2695高效液相色谱仪,2489紫外检测器(Waters公司);XP205电子天平(上海梅特勒有限公司);DT255 超声清洗机(BANDELIN公司)。银黄口服液,爱迪森(北京)生物科技有限公司提供,100 mL/瓶,批号1207271、1212031、1212211);绿原酸对照品(中国兽医药品监察所,批号Z0260611,含量96.6%);黄芩苷对照品(中国兽医药品监察所提供,批号Z0271109,含量98.8%)。HPLC 级乙腈,磷酸等其他试剂为分析级。
2 方法与结果
2.1 色谱条件 色谱柱Discovery C18柱(4.6×150 mm,5 μm),检测波长327 nm,进样体积10 μL,以乙腈为流动相A,以0.4%磷酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速1 mL/min。理论板数按绿原酸峰计算应不低于2 000,按黄芩苷峰计算应不低于2 500。见表1。

表1
2.2 溶液制备
2.2.1 绿原酸对照品贮备液 精密称取绿原酸对照品10.38 mg,置100 mL量瓶中,加50%甲醇溶解后稀释至刻度,摇匀,作为绿原酸贮备液。
2.2.2 黄芩苷对照品贮备液精密称取黄芩苷对照品10.20 mg,置100 mL量瓶中,加甲醇超声溶解并稀释至刻度,摇匀,作为黄芩苷贮备液。
2.2.3 混合对照品溶液 分别精密量取绿原酸贮备液2 mL和黄芩苷贮备液25 mL,置50 mL量瓶中,加水稀释至刻度,摇匀,即得。
2.2.4 样品溶液 精密量取样品1 mL,置50 mL棕色量瓶中,加50%甲醇稀释至刻度,摇匀;精密量取3 mL,置25 mL棕色量瓶中,加50%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
2.3 测定法 分别精密吸取混合对照品溶液与样品溶液各10 μL,注入液相色谱仪,测定。数见图1、图2。
2.4 线性关系考察 分别精密量取绿原酸贮备液和黄芩苷贮备液适量,加50%甲醇稀释制成一系列标准溶液,含绿原酸浓度分别为1.00、3.00、5.01、7.02、10.02、20.05 μg/mL,含黄芩苷浓度分别为10.07、30.03、50.39、70.54、100.78、201.55 μg/mL。按上述色谱条件进样测定以对照品溶液质量浓度(μg/mL)为横坐标,峰面积为纵坐标,绘制标准曲线。绿原酸线性方程:Y=3.123 0×10-5X+0.034 0,R2=0.999 9,绿原酸浓度范围在1.00~20.05 μg/mL,见图1。黄芩苷线性方程:Y=5.047 9×10-5X-0.039 6,R2=0.999 9,黄芩苷浓度范围在10.07~201.55 μg/mL,见图3、图4。
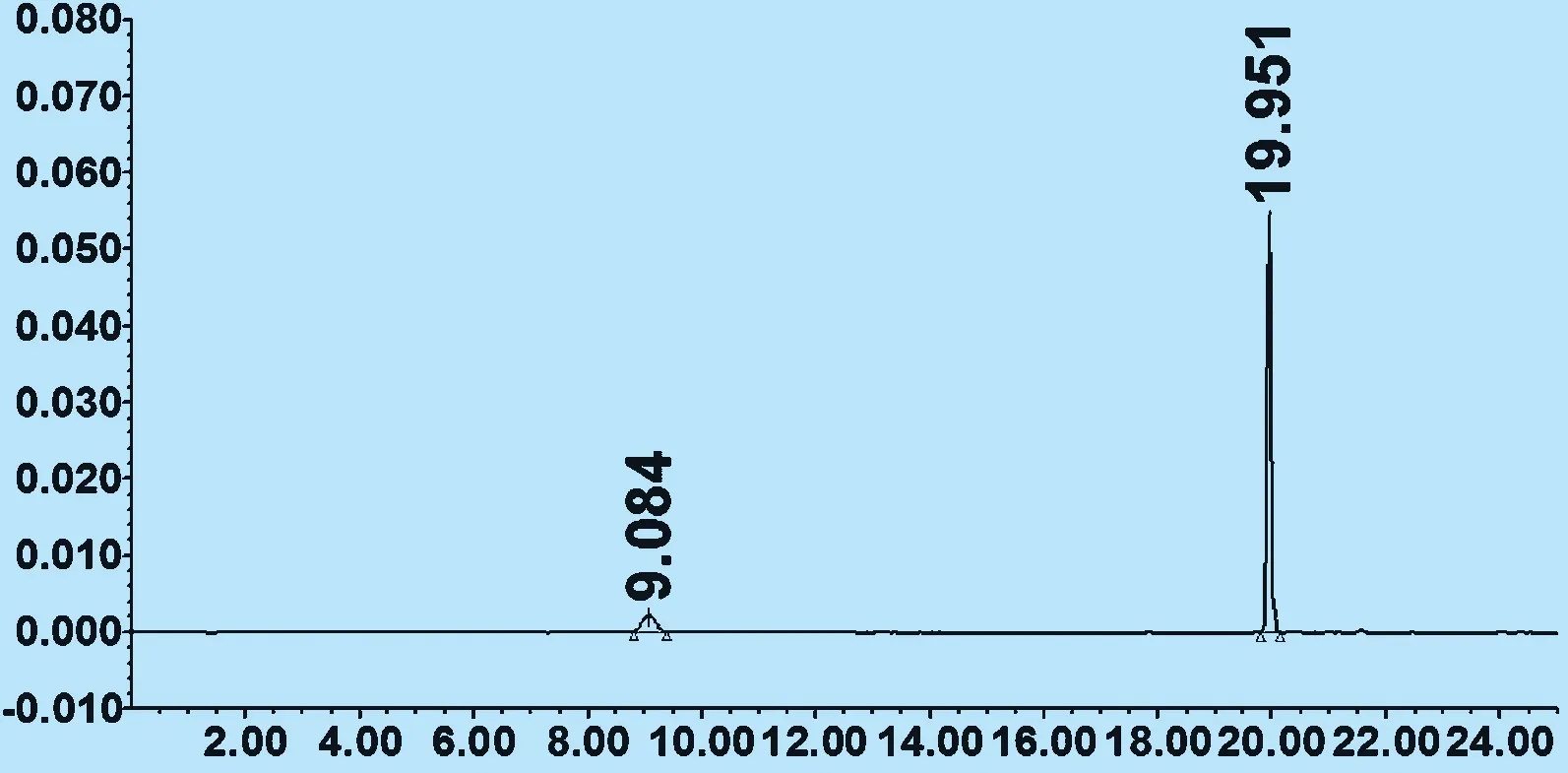
图1 混合对照品
注:绿原酸峰的保留时间为9.084 min,黄芩苷峰的保留时间为19.951 min
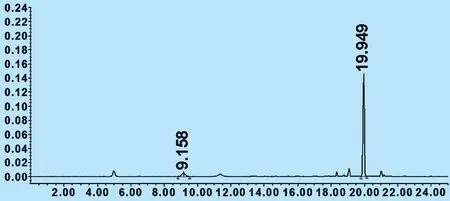
图2 供试品
注:绿原酸峰的保留时间为9.158 min,黄芩苷峰的保留时间为19.949 min
2.5 精密度考察 分别精密量取绿原酸和黄芩苷对照品溶液,按照2.1项下条件连续进样6次,绿原酸和黄芩苷峰面积的标准偏差(RSD)分别为0.76%,0.65%。
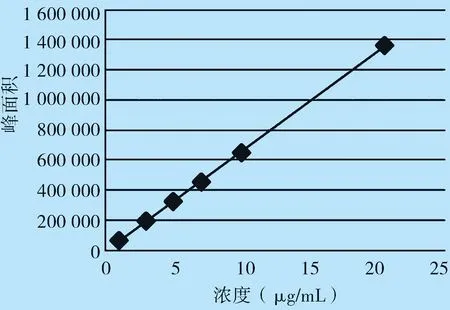
图3 绿原酸线性图
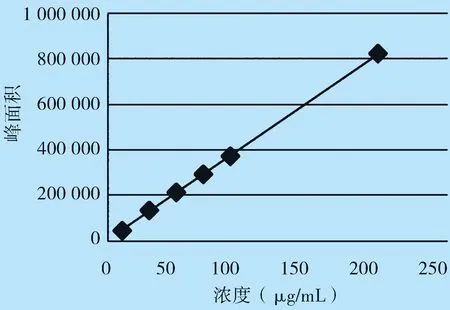
图4 黄芩苷线性图
2.6 重复性考察 取同一批号样品,按2.3项下方法制备供试品溶液6份,按2.1项下色谱条件进样测定,计算峰面积的相对标准偏差(标准规定应不大于2.0%)分别为0.13%和0.32%。
2.7 稳定性考察 精密吸取同一批银黄口服液供试品溶液,按照上述色谱条件,30 h内分别每隔2 h进样一次,记录峰面积,计算相对标准偏差(RSD)。数据见表2。
2.8 加样回收试验 本加样回收试验按照80%、100%、120%的量进行添加回收,即每个浓度制备3份平行样,分别平行进样3针。精密量取已知含量的银黄口服液9份,每份1 mL,置100 mL棕色量瓶中,分别按照绿原酸和黄芩苷实际含量的80%、100%、120%精密加入绿原酸和黄芩苷,用水稀释至刻度,摇匀;精密量取3 mL,置25 mL棕色量瓶中,用50%甲醇稀释至刻度,摇匀,按2.1项下色谱条件进样测定,计算回收率及相对标准偏差(RSD)。所得数据见表3、表4。
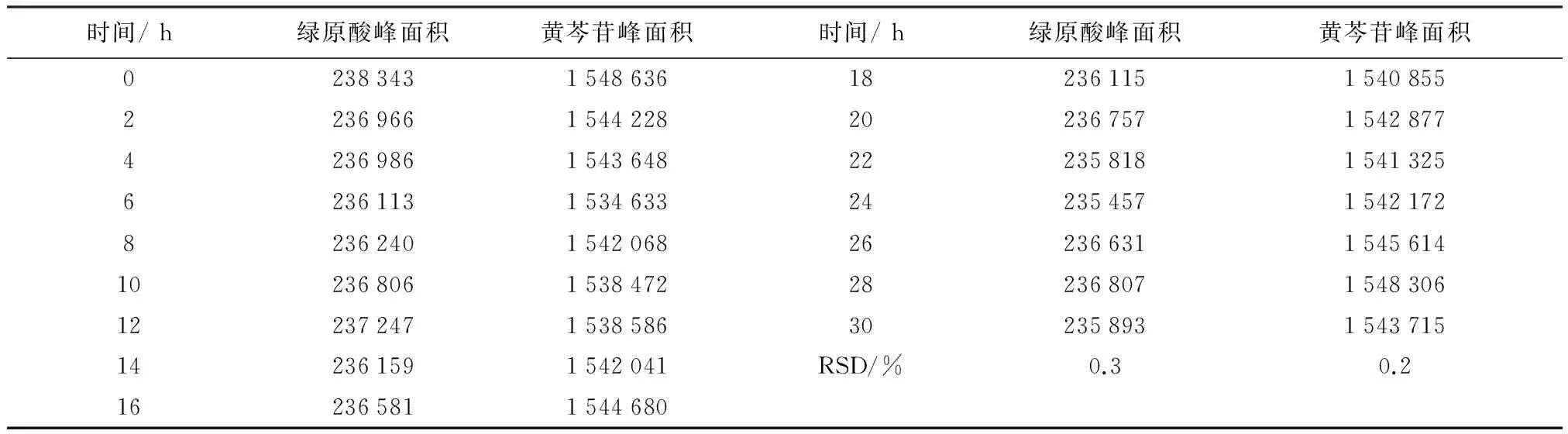
表2 新方法溶液稳定性考察
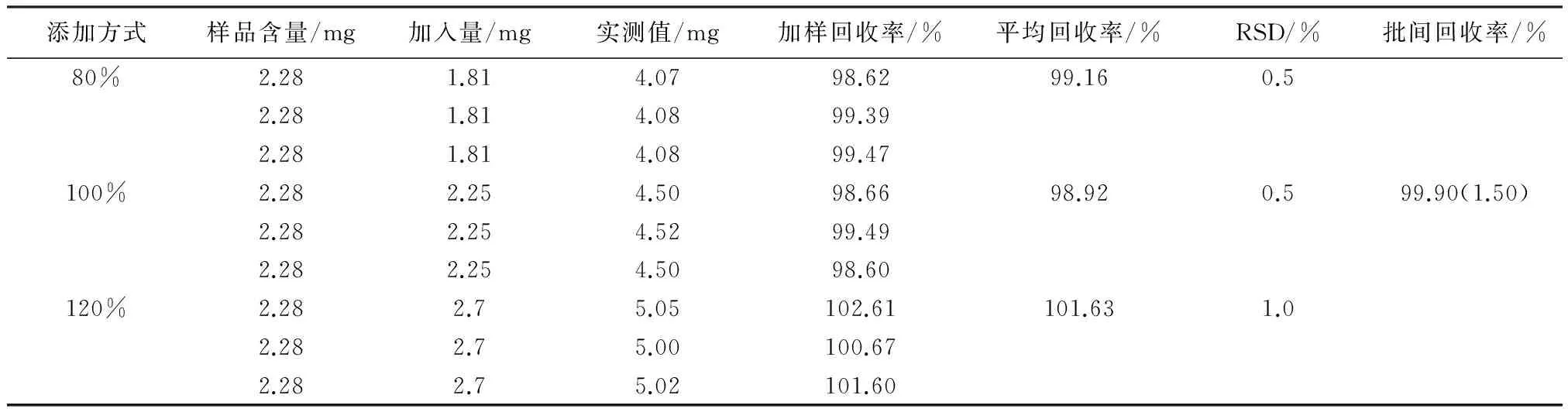
表3 绿原酸加样回收试验结果 (n=9)
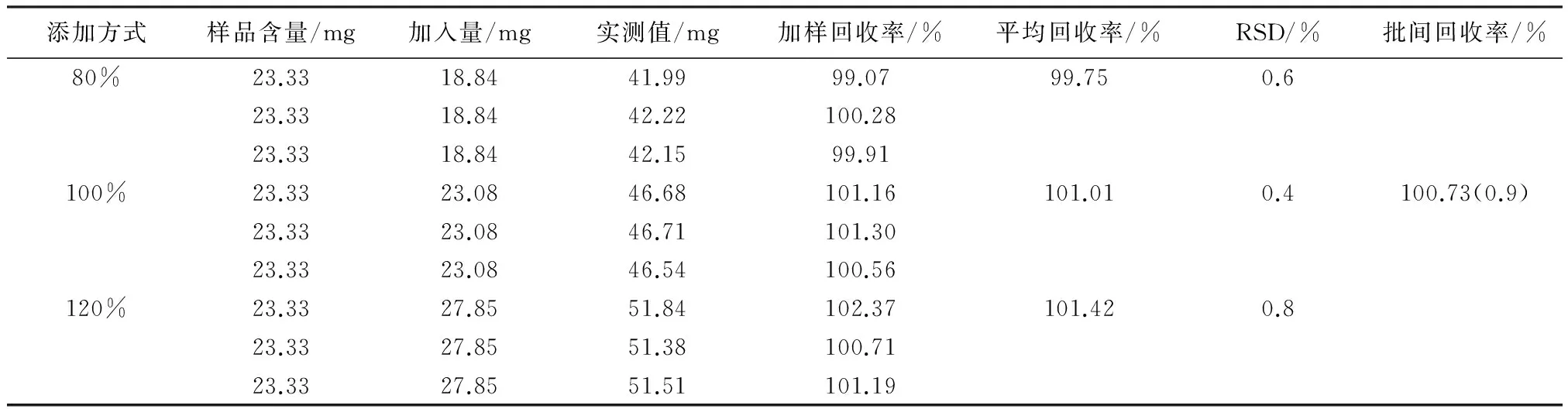
表4 黄芩苷加样回收试验结果 (n=9)
2.9 色谱柱耐用性考察 分别采用不同的高效液相色谱仪和色谱柱,按照新方法色谱条件,分别精密吸取绿原酸和黄芩苷的混合对照品溶液、样品溶液注入色谱仪测试。结果见表5。
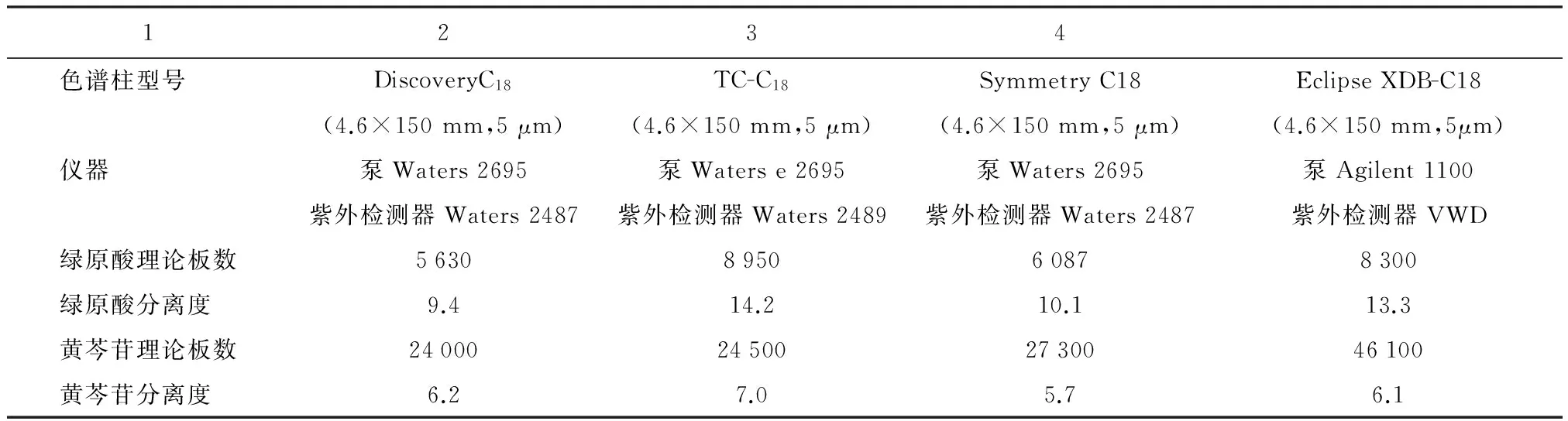
表5 色谱柱耐用性考察
3 讨论
3.1 样品的制备 样品溶液按照原方法进行制备,由于黄芩苷和绿原酸浓度相差10倍左右,本研究将绿原酸对照品溶液浓度降低10倍,以保持绿原酸良好的线性关系。选择溶解供试品溶剂时,本研究考察了采用50%甲醇或水作为第一步的溶剂,结果显示含量无差异,说明50%甲醇或水对银黄注射液均具有良好的溶解性,考虑对照品溶液的溶剂为50%甲醇,所以最终选择50%甲醇作为第一步溶解的溶剂。
3.2 流动相的选择 原质量标准草案中,绿原酸和黄芩苷的等度洗脱流动相不同,比例相差亦较大,为了采用同一种流动相同时对两者进行含量测定,本研究参考文献研究及两者流动相溶剂和比例的基础上[7-9],有机相选用洗脱能力较强的乙腈进行梯度洗脱。洗脱时,先大比例水相分离出绿原酸,然后再提高有机相比例洗脱出黄芩苷,直至黄芩苷完全流出以及再无其他组分干扰后,逐渐过渡到最初比例平衡色谱柱,循环运行下一针。
3.3 波长选择 文献报道中[10-11],黄芩苷检测波长一般为274 nm,绿原酸检测波长以327 nm居多。银黄口服液中绿原酸含量远低于黄芩苷,为了得到目标峰美观的色谱图,本方法选择测定绿原酸的波长为绿原酸和黄芩苷同时测量的波长对两者成分进行同时检测。
4 结论
通过本方法所得目标成分分离度好、理论塔板数高,方法稳定可靠,可作为银黄口服液的含量测定方法。实际检测中,此方法操作相对简单、快捷,检测时间为较原方法缩短1 倍以上,大大提高了检测效率,对促进实际检测和生产具有积极的意义。
[1] 陈美娟,葛李,顾立,等.银黄口服液抗菌作用研究[J].时珍国医国药,2007,18(1):107-108.
[2] 中国兽药典委员会.《中华人民共和国兽药典》2012 年版2部[S].
[3] 张静,刘文翔,韩艳婷.HPLC 法测定银黄口服液中绿原酸、黄芩苷和木犀草苷的含量[J].西北药学杂志,2011,26(4):237-239.
[4] 桑旭峰,吴海霞,徐奇超.HPLC 同时测定银黄口服液中绿原酸和黄芩苷含量[J].中成药,2005,27(1):96-98.
[5] 孙博,李继昌,王小飞,等.鸡血浆中黄芩苷及绿原酸的HPLC 法建立[J].中国兽药杂志,2013,47(3):31-33.
[6] 翟淑平,黄斌,王秀敏,等.HPLC 法同时测定银黄可溶性粉中绿原酸和黄芩苷的含量[J].中国兽医杂志,2006,26(12):1880-1882.
[7] 赵秀香,李启艳,王磊.H PLC 法测定银黄注射液中绿原酸的含量[J].药物分析杂志,2006,26(12):1880-1882.
[8] 于加平.HPLC 法测定徐长卿中绿原酸的含量[J].中国兽药杂志,2012,46(1):34-36.
[9] 王丽聪,曹玉华,徐江兰,等.银黄口服液含量的质量控制及其高效液相色谱指纹图谱的研究[J].色谱,2006,24(4):367-372.
[10] 韦国兵,胡奇军,廖夫生.HPLC 法测定银黄口服液中黄芩苷的含量[J].江西师范大学学报(自然科学版),2013,37(5):529-531.
[11] 朱啸风,孙钦美,刘景俊.银黄口服液中黄芩苷和绿原酸含量的测定[J].齐鲁药事,2004,23(6):25-26.
Study on the Improvement of determination of chlorogenic acid and baicalin in Yinhuang Oral Liquid by HPLC
ZHOU Yan-fei1, LI Han1, WANG Ya-fang1, LI Ying-chao1, WEI Lei1, WANG Hai-jun2,3
(1.Beijing Institute of Veterinary Drug Control,Beijing 102629,China;2.Beijing Centre Biology Co.,Ltd,Beijing 102206,China;3.Beijing Veterinary Drugs Engineering Research Center,Beijing 102206,China)
To establish a HPLC method for simultaneous determination of chlorogenic acid and baicalin in Yinhuang oral liquid,the Discovery C18(4.6×150 mm,5 μm)column was adopted with acetonitrile and 0.4% phosphoricacid solution was as the mobile phase and gradient elution.The flow rate was 1.0 mL/min-1,the volume of injection was 10 μL,and the detective wavelength was set at 327 nm.There were better linear relationships between the peak area and concentration at the range of 0.5~20 μg/mL(R2=0.999 8),5~199 μg/mL(R2=0.999 8)for chlorogenic acid and baicalin,respectively.The average recovery rates of chlorogenic acid and baicalin were 99.90% and 100.73%,respectively,and the RSD was less than 1.0%.The HPLC method is reproducible,specific,accurate,and can be used as a quantitative analysis of chlorogenic acid and baicalin in Yinhuang oral liquid.
Yinhuang Oral Liquid ; HPLC ; baicalin ; chlorogenic acid
WANG Hai-jun
2015-08-31
周艳飞(1972-),女,高级兽医师,大专,研究方向为兽药质量控制,E-mail:1956437829@qq.com
王海军,E-mail:wtianlong86@126.com
R927.2
A
0529-6005(2016)10-0089-04
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
化工管理(2020年18期)2020-07-15
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年7期)2018-08-21
中成药(2017年10期)2017-11-16
人民周刊(2016年11期)2016-06-30
中国猪业(2016年1期)2016-01-28
中国医药生物技术(2014年4期)2014-01-23
