
77次体外诊断试剂生产企业体系考核766条缺陷项的原因分析
2016-12-15 00:40吴林松何蕊
中国医疗器械信息 2016年7期
吴林松何蕊
1 湖州市市场监督管理局 (湖州 313000)
2 浙江省医疗器械审评中心 (杭州 310009)
77次体外诊断试剂生产企业体系考核766条缺陷项的原因分析
吴林松1何蕊2
1 湖州市市场监督管理局 (湖州 313000)
2 浙江省医疗器械审评中心 (杭州 310009)
目的:通过对体外诊断试剂生产企业体系考核中缺陷项的原因分析,为体外诊断试剂生产企业今后生产中需要注意的事项以及检查员应关注的方向提供参考。方法:实行《体外诊断试剂生产企业质量体系考核实施规定(试行)》之后对被检查企业的缺陷项进行统计分析。结果:77次体外诊断试剂生产企业体系考核766条缺陷项,涉及127个条款,占82%。结论:关注缺陷项的原因分析,能够有效控制体外诊断试剂产品的质量,不断提高体系考核的通过率和检查员的工作效率。
体外诊断试剂生产 体系考核 缺陷项 分析
吴林松,主管药师
自国食药监械[2007] 239号《体外诊断试剂生产企业质量体系考核实施规定(试行)》(以下简称《规定》)实行以来,浙江省药品认证中心(以下简称中心)依据《规定》对32家体外诊断试剂生产企业的77次考核进行了调研,共计缺陷项766个,涉及127个条款,占总条款82%。对其中出现频率较高的5个环节(设施、设备与生产环境控制、采购控制、生产过程控制、检验与
质量控制,附录A中体外诊断试剂生产用净化车间环境与控制要求)的缺陷项进行了原因分析,中心希望通过对缺陷项的原因分析,能给体外诊断试剂生产企业在今后生产中需要注意或改进的事项以及检查员在体系考核中应关注的方向提供参考。
1.资料与方法
1.1 考核依据
《体外诊断试剂生产企业质量体系考核实施规定(试行)》。
1.2 考核时间
2007年5月~2013年07月。
1.3 考核对象
浙江省食品药品监督管理局管辖范围内提出体系考核申请的32家体外诊断试剂生产企业。1.4考核内容
对组织机构、人员与质量管理职责;设施、设备与生产环境控制;文件与记录;设计控制与验证;采购控制;生产过程控制;检验与质量控制;产品销售与客户服务控制;不合格品控制、纠正和预防措施;不良事件、质量事故报告制度;附录A 体外诊断试剂生产用净化车间环境与控制要求等环节进行考核,并记录缺陷项。
1.5 方法
对缺陷项出现频率15次以上的条款进行列表和数据统计分析。
2.结果
32家体外诊断试剂生产企业的设施、设备与生产环境控制、采购控制环节等5个环节的缺陷项情况分别见表1至表5。
2.1 设施、设备与生产环境控制环节缺陷项情况,见表1。
2.1.1 分析与讨论
表1中,设施、设备与生产环境控制这章节共出现缺陷项178次,在体系考核过程中涉及的内容面较广,出现问题也较多。分析原因是相关的保管和操作人员缺乏培训或培训工作不到位,操作规范习惯的养成和意识差,工作原理不吃透等情况。所以我们对以下环节做重点要求和关注:(1)厂房、设施与设备应与体外诊断试剂生产规模相适应;(2)生产、研发、检验等区域应相互分开;(3)原料、辅料、包装材料、半成品、成品等各个区域必须划分清楚,防止出现差错和交叉污染;(4)所有物料的名称、批号、有效期和检验状态等标识必须明确,台帐应清晰明确,做到帐、卡、物一致;(5)易燃、易爆、有毒、有害、具有污染性或传染性、具有生物活性或来源
于生物体的物料其存放应符合国家相关规定,应做到专区存放并有明显的识别标识,由专门人员负责保管和发放;(6)生产过程中所涉及的化学、生物及其他危险品,企业应列出清单,并制定相应的防护规程,其环境、设施与设备应符合国家相关安全规定;(7)对设备的有效性定期进行验证;(8)对各类物料的仓储环境及控制应符合规定储存,并进行定期监测和记录;(9)保管和操作人员的培训情况等。企业如果对这些细节进行重点关注并整改到位,将会大步减少缺陷项出现的数量。

表1. 施、设备与生产环境控制环节缺陷项情况
2.2 采购控制环节缺陷项情况,见表2。
2.2.1 分析与讨论
采购控制这个章节中出现缺陷项59条,企业的采购部门和人员往往对供货商的送货时间及价格关注较多,而对供货方的资质评价、供需合同中物料的技术指标和质量要求容易疏忽。众所周知,原材料(包括校准品、质控品)的采购过程是否规范是企业生产工艺稳定和产品质量的重要保障,所以采购部门要确实履行职责,严格按采购控制文件要求执行,完善对供货方的资质评估工作,明确物料的技术指标和质量要求并体现在供需合同中,并按以上要求进行进货检验和资料的留存。
2.3 生产过程控制环节缺陷项情况,见表3。
2.3.1 分析与讨论
生产过程控制中出现缺陷项122条,主要问题在生产过程中使用的设备和仪器状态的有效性缺失,生产过程中一些记录和验证工作不完整,物料平衡和环境监测控制工作不到位。因此,企业应按照批准的工艺进行生产,应按制定生产所需的工序流程、程序文件和标准规程操作,明确关键工序或特殊工序,确定质量控制点,并规定和留存生产记录。应明确每批产品中关键物料的平衡关系,并对每批产品中关键物料进行物料平衡核查,应明确物料平衡超出可接受标准的处理方法。不同批次产品生产工序结束后必须进行清场,确认合格后才可以入场进行其他批次产品的生产,企业应保存清场记录。清场时,配制和分装器具等应当进行清洗、干燥等洁净处理,并进行验证,器具清洗干燥后应有有效保存时间及相关验证。
2.4 检验与质量控制环节缺陷项情况,见表4。
2.4.1 分析与讨论

表2. 采购控制环节缺陷项情况
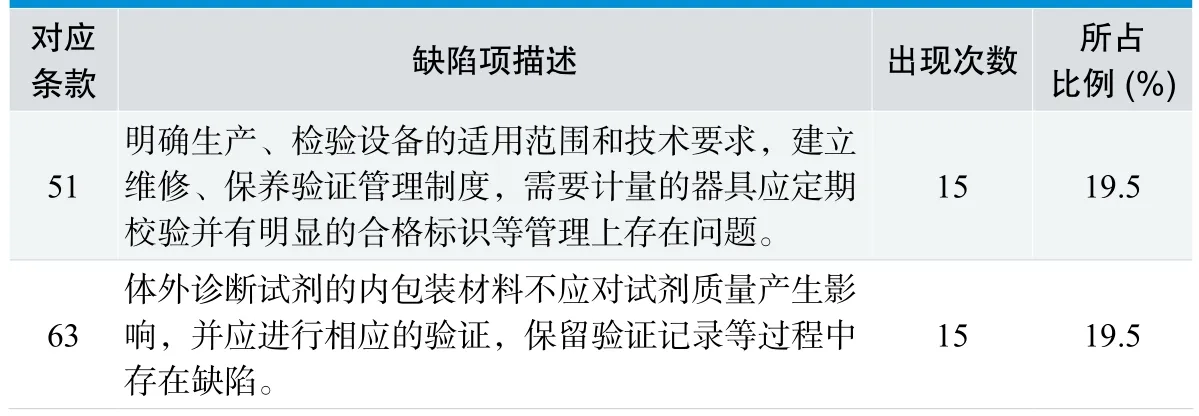
表3. 生产过程控制环节缺陷项情况
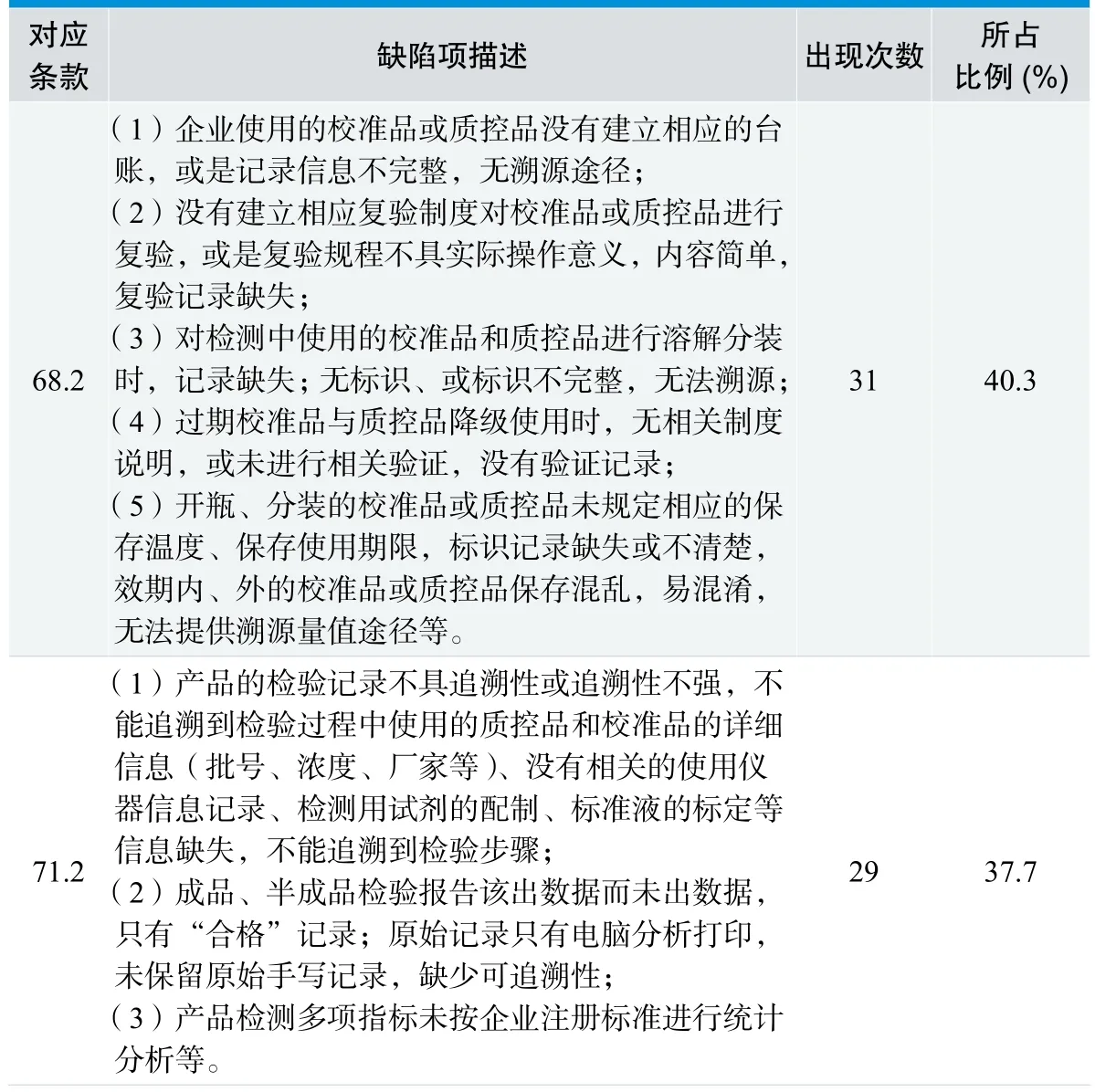
表4. 检验与质量控制环节缺陷项情况
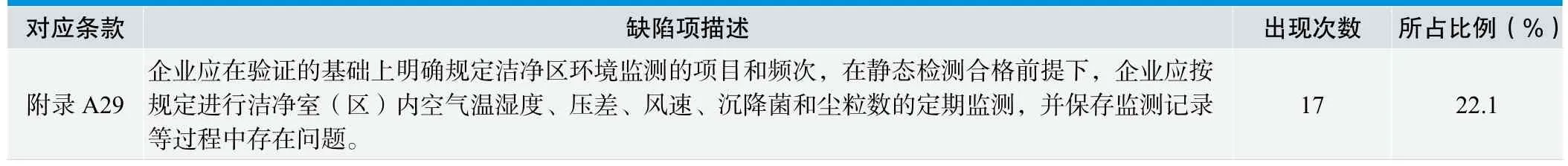
表5. 附录A 体外诊断试剂生产用净化车间环境与控制要求环节缺陷项情况
表4中出现缺陷项128条,其中68.2条款:对检测中使用的校准品和质控品应建立台帐及使用记录;应记录其来源、批号、效期、溯源途径(或靶值转换方法)、主要技术指标、保存状态等信息;应定期复验其性能并保存记录等该条款在77次考核中出现的比例高达40.3%。主要原因是操作人员较注重操作过程,而对操作步骤和信息的记录相对不全,特别是对使用过程中的校准品、质控品和配制的试剂标识不够重视,应注明的详细信息(批号、浓度、厂家、配置时间等)不全,没有相关的使用仪器信息记录,校准品、质控品等复验制度落实不到位,给生产和检验结果的追溯和可靠性增加了难度和风险。田少雷也曾报道体外诊断试剂质量管理体系中最为薄弱的环节是文件与记录控制,对文件与记录的不重视必然会影响到质量管理体系的各主要环节的正常运行。[2]
2.5 附录A 体外诊断试剂生产用净化车间环境与控制要求环节缺陷项情况,见表5。
2.5.1 分析与讨论
附录A中体外诊断试剂生产用净化车间环境与控制要求的内容在本次调研中出现频率也是较高的,共出现116次。其中附录A29出现17次,占22.1%,给企业的体系考核通过增加了难度。主要原因是企业对车间一线生产人员的操作和生产过程的质量控制相对重视,而对净化车间清洁人员、空调机组和制水间操作人员的管理工作不到位,人员意识跟不上体系要求,往往只知其然,而不知所以然。所以对仪器仪表的数据记录积极性不高、责任心不强(不知道这些数据的含义和意义),出现一些异常问题也不能发现和分析,更不知道如何解决。我们体系考核要求企业全员意识的到位,不是某个部位或者是某个人的意识有了就行了。
3.存在问题的主要原因
人员意识的问题是企业亟需改正的首要任务,存在问题的原因主要有:(1)首先是企业负责人或法人代表对法律法规和相关标准不熟悉。相当一部分企业负责人未进行法律、法规及ISO13485标准的培训,有的培训了只是拿个证书,未对培训的内容在企业生产质量管理体系过程中进行体会和消化,轻质量重效益,思想意识上与体系的要求仍存在差距,而对企业生产质量管理体系是否有效运行认为只是管理者代表和几个负责人的事情,体系管理水平对产品质量的重要性未引起高度注意,缺乏防患于未然的大局观念,没有意识到一旦产品出现质量状况会给企业带来灾难性打击问题的严重性。(2)管理层人员过于服从老总的意愿,在质量与利润的天平秤上偏向了利润,服从与原则的问题上更趋于服从,一些负责人的责任心和原则性不强。其中也有一些企业委托代办公司申请或指导质量体系,所以现场提出许多问题时岗位上的操作人员与管理者回答的结果不一样。其实请人指导应该是好事,可以让企业较快的掌握和理解体系的精神,但一定要企业全体员工充分领会体系的用意并运行到工作中去,殊不知产品的质量是企业的生命,如果这方面不能引起足够重视,企业的产品还会存在这样那样的安全隐患。(3)车间员工的培训与标准化操作能力不同步。洁净车间员工微生物知识培训和岗位操作的继续再教育不到位,基层员工接受知识的
能力偏低是普遍情况,但越是如此就越要更加重视基层员工的培训,充分吸取短板原理的教训,重视企业整体的生产质量管理水平。
4.结语
综上所述,对体外诊断试剂生产企业体系考核中缺陷项的关注和控制,是企业保证产品质量、降低生产成本和提高检查通过率的一个重要环节。尤其是缺陷项跟踪改正措施的有效性和举一反三解决问题能力的提高,往往会起到事半功倍的效果。所以,企业在每次体系内审过程中针对每一缺陷项的整改和跟进措施要记录、分析,应及时进行状态的更新,有效控制产品的质量,不断完善体系的科学性和有效性。同时通过缺陷项的原因分析也是提高检查员能力的快速途径。
质量管理规范的检查侧重的是企业在质量体系管理是否存在缺陷,而不是是否满足检查条款的要求,在检查过程中不用重点考虑某个缺陷项应该具体归属到哪个检查条款中去,更多的要考虑是否满足《医疗器械生产质量管理体系规范》总则的要求。企业为自己的产品质量负责,从产品风险着手,建立质量体系,监督部门专心做好监督检查。[3]
[1]《体外诊断试剂生产企业质量体系考核实施规定(试行)》[P]2007-04-28.
[2]田少雷.对我国体外诊断试剂质量体系考核中发现问题的统计分析[J]中国医疗器械杂志,2012,36(6):433-437
[3]高琳琳. 医疗器械企业如何建立有效的质量管理体系[J].科技信息(科技教育版),2006,2:100
Cause Analysis of 766 Deficiency during the Evaluation of the Manufacturer of 77 In Vitro Diagnosis Reagent
WU Lin-song1He Rui2
1 Market Supervision Administration of huzhou Municipality (Huzhou 313000)
2 Zhejiang Province Medical Devices Evaluation Center (hangzhou 310009)
Objective: By analyzing the cause of deficiency during the evaluation of the manufacturer of 77 In Vitro Diagnosis(IVD) reagent, provide the IVD reagent manufacturer and inspector with reference on points for attention. Methods: Statistical analysis on the deficiency of inspected manufacturer after the execution of Regulation of Quality Evaluation of IVD Reagent Manufacturer (Test). Results: Among the 766 deficiencies evaluated on 77 IVD reagent manufacturer, 127 causes is covered, accounting for 82%. Conclusion: Focus on the analysis of causes for deficiency, improve the quality of IVD reagent, increase the percent of pass of system evaluation and efficiency of inspector.
IVD reagent manufacturer, system evaluation, deficiency, analysis
1006-6586(2016)04-0021-04
R194
A
2015-02-11
猜你喜欢
机械工业标准化与质量(2022年8期)2022-10-09
现代仪器与医疗(2021年1期)2021-06-09
小学生学习指导(高年级)(2021年3期)2021-04-06
保健文汇(2020年8期)2020-12-02
中国特种设备安全(2019年11期)2020-01-16
今日农业(2019年16期)2019-09-10
中国资源综合利用(2017年4期)2018-01-22
软件(2015年9期)2015-12-25
船舶标准化工程师(2015年5期)2015-12-03
中国卫生(2014年11期)2014-11-12
