
国内外医疗器械分类管理思路和规则的对比分析
2016-12-15 00:40周良彬伍倚明李伟松张春青
中国医疗器械信息 2016年7期
周良彬伍倚明李伟松张春青
1 广东省医疗器械质量监督检验所 (广州 510663)
2中国食品药品检定研究院 (北京 100050)
国内外医疗器械分类管理思路和规则的对比分析
周良彬1伍倚明1李伟松1张春青2
1 广东省医疗器械质量监督检验所 (广州 510663)
2中国食品药品检定研究院 (北京 100050)
我国新版《医疗器械分类规则》于2015年7月发布,自2016年1月正式实施。为了促进深入理解和掌握医疗器械分类规则,本文汇总、整理、分析了欧盟、美国、日本等国家/地区的相关法规文件,分析了各国分类管理思路,对比了各国分类规则的异同。
医疗器械 分类 规则 管理思路
0.引言
目前,我国正在进行医疗器械分类管理的深化改革。2014年3月7日,我国《医疗器械监督管理条例》[1](中华人民共和国国务院第650号,以下简称《监管条例》)正式发布,其中明确规定“国家对医疗器械按照风险程度实行分类管理”(第四条)。2014年5月30日,在继《监管条例》之后,国家食品药品监督管理总局(以下简称“食药监局”)在对2002版《医疗器械分类目录》[2]中第一类产品进行整理、补充、完善的基础上,发布了《第一类医疗器械产品目录》[3]。2015年
7月14日,新版《医疗器械分类规则》[4](国家食品药品监督管理总局令第15号,以下简称《分类规则》)发布,于2016年1月1日正式实施。
在本次《分类规则》的修订说明中介绍,本次新版文件的修订参考并容纳了欧盟、国际医疗器械监管机构论坛(IMDRF)指南等有关分类的资料,表明在经济全球化的今天,作为医疗器械进出口大国,我国的医疗器械分类管理体系在立足于国情的同时,也受到了国际监管政策的影响。
为了便于监管部门、企业等相关人员了解各国分类监管思路,本文对我国、欧盟、美国、日本、加拿大、新加坡等医疗器械分类管理法规做一简介,并对其思路和规则进行对比分析,以供参考。
1.各国(地区)分类管理法规体系基本情况介绍
由于各国(地区)医疗器械监管法律体系存在差异,与之相适应的分类管理法规体系也就呈现出不同特点。以下做简要介绍:
1.1 我国医疗器械分类管理法规体系
我国的分类管理法规体系是以《监管条例》为基础,以《分类规则》为指导,具体体现为《医疗器械分类目录》。
根据《监管条例》[1],我国医疗器械按照风险从低到高分为第一类、第二类、第三类,对应这三个管理类别,相应的主要管控措施为:第一类医疗器械实行产品备案管理,向所在地市级食药监部门提交资料;第二类、第三类医疗器械实行产品注册管理,其中第二类产品注册,向所在地省级食药监部门提交资料,第三类产品注册,向国务院食药监部门提交注册申请资料。
近几年,为了配合医疗器械监管全面改革,食药监总局不断深化医疗器械分类管理体制改革,组织专项工作对《监管条例》、《分类规则》、《分类目录》进行修订,并着手组建专门的医疗器械分类专家组织。目前,新版的《监管条例》、《分类规则》已分别于2014、2015年发布,医疗器械分类技术委员会也于2015年11月宣告成立。
1.2 欧盟医疗器械分类管理法规体系
欧盟的医疗器械监管相关法令主要有三个,分别应用于有源植入医疗器械(AIMD,90/385/ EEC[5])、普通医疗器械(MD,93/42/EEC[6])和体外诊断医疗器械(IVDMD,98/79/EC[7])。
有源植入医疗器械属于高风险,按最严的措施进行管控。
普通医疗器械指令[6]中,采用18条分类规则将器械按照风险由低到高分为I、IIa、IIb和III类。在此基础上,为了便于对分类规则的理解和使用,欧盟发布了指令93/42/EEC的应用指导原则“医疗器械分类”[8],对相关概念和规则进行进一步的阐述和举例说明。
和普通医疗器械分类思路不同,目前指令98/79/EC[7]对于体外诊断医疗器械的分类采用了List(清单)的形式,对不同风险特征的产品进行了区分,在该指令Article 9 Conformity assessment procedures(符合性评估程序)中将产品划分为属于ANNEX II中List A、List B以及不属于List的产品,并提出了对应的符合性评估要求。
近年来,欧盟为加强医疗器械的准入和监管,于2012年提出新的法规提案对93/42/EEC等三个主要的指令进行了调整和修改。新的提案将有源植入医疗器械(90/385/EEC)和普通医疗器械(93/42/EEC)的指令进行了合并,形成了一个新的法规[9](Regulation,由Directive到Regulation,法律层级上升),其中Annex Ⅶ Classification criteria对分类标准进行了阐述,分类规则增至21条。
与之同步,体外诊断医疗器械(IVDMD)方面,其监管文件同样从指令(Directive)上升到了法规(Regulation)[10],其中关于IVDMD分类思路也发生了重大改变,将之前的清单改为基于规则的分类体系,利用7条分类规则将IVDMD
类产品依据风险从低到高分为A、B、C、D四个管理类别。
1.3 美国医疗器械分类管理法规体系
在美国《食品、药物与化妆品法案》[11]第513条“人用器械的分类”中,根据保障产品安全、有效所需采用措施的不同,将人用器械分为Ⅰ、Ⅱ、Ⅲ三类,分别采用一般控制、特殊控制、上市前批准审批的措施进行管理。
和我国、欧盟都采用分类规则指导医疗器械分类不同,美国FDA没有医疗器械分类规则,而是依据风险分析的基本原则,对各种产品进行逐个具体分析,确定产品管理类别。美国FDA将所有医疗器械(含IVD)按照一定原则划分为19个医学专业类别(medical specialty),每个专业类别项下,按照产品的使用目的或该类产品的特性等,再分为若干子类别,子类别下规定具体的
产品种类,每个产品种类项下包含有编号、名称、定义、分类、管控措施等内容。对19个医学专业类别的表述可在美国联邦规章典(Code of Federal Regulations,CFR)[12]中找到,分别为:麻醉学、心血管、牙科、耳鼻喉、胃肠病学和泌尿科、一般及整形外科、一般医院用品、神经病学、妇产科、眼科、整形外科、物理医学、放射学/成像、临床化学、临床毒理学、病理学、血液学、免疫学、微生物学。这些类别分别划入了CFR从Part862到Part892共16个文件中,其中临床化学和毒理学、血液学和病理学、免疫和微生物学分别合并为CFR Part 862、864、866。
为确保分类体系的实用性、可操作性,FDA对这16个CFR文件中的1800个产品种类进行了进一步细分,建立了包含6000多个具体产品品种的分类数据库(Product Classification Database),该数据库每周更新,作为网上公开数据库,供公众查询。
1.4 日本医疗器械分类管理法规体系
日本医疗器械管理的基本法为《药事法》,而医药器械、体外诊断试剂进行等级分类规则是按照2013年5月10日厚生劳动省医药食品局长通知的《高度管理医疗器械、管理医疗器械以及一般医疗器械有关的等级分类规则修改》,通过厚生劳动省大臣告示的方式让业界了解。
按照医药品医疗器械等法律[13]的规定,日本根据产品发生问题对人体影响程度的差异将医疗器械分为Ⅰ、Ⅱ、Ⅲ、Ⅳ共四个等级。等级Ⅳ和等级Ⅲ医疗器械为高度管理医疗器械,等级Ⅱ医疗器械为管理医疗器械,等级Ⅰ医疗器械为一般管理医疗器械。其中,高度管理需要PMDA(pharmaceuticals and medical devices agency,日本药品和医疗器械管理机构)审查,厚生省大臣认可;管理需要第三方认可;一般管理则备案即可。
体外诊断试剂则根据诊断信息风险,进行等级分类,分为Ⅰ、Ⅱ、Ⅲ共三个等级。
具体的分类实践中,厚生省综合参考GHTF分类规则进行的分类和现有分类的两方面信息,建立了JMDN数据库,其中包含名称、定义、管理类别、管控措施等方面内容。该数据库由厚生劳动省医药食品局进行维护更新。
1.5 加拿大医疗器械分类管理法规体系
加拿大医疗器械监管法规《Medical Devices Regulations》[14](SOR/98-282,最后一次修订时间为2011-12-16)中规定了分类管理的相关内容,其医疗器械采用基于风险的分类规则系统,其中非IVD医疗器械分类规则16条,IVD器械分类规则9条,将器械按照风险水平从低到高分为Ⅰ、Ⅱ、Ⅲ、Ⅳ共四个类别。为了帮助各方面深入了解其分类体系,加拿大卫生部的健康产品与食品管理部门发布指导文件《Guidance on the Risk-based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs)》(2015-04-23发布)对非IVD医疗器械分类规则进行解释和举例说明。
1.6 IMDRF(GHTF)医疗器械分类文件
作为医疗器械法规的国际协调组织,国际医疗器械监管者论坛(International Medical Device Regulators Forum,IMDRF)的前身——全球协调工作组(The Global Harmonization Task Force,
GHTF)发布了指导文件《Principles of medical devices classification》[15](2012)、《Principles of in vitro diagnostic (ivd) medicaldevicesclassificatio n》[16](2008),作为各国建立其分类管理系统的基础。欧盟、日本、新加坡都不同程度地采用了GHTF的指导原则。
GHTF的指导文件中通过17条规则[15],将非IVD的医疗器械依据危险从低到高分为A、B、C、D四个管理类别;IVD医疗器械则是以7条规则[16],同样分为四类。
2.国内外非IVD医疗器械分类规则对比
需要说明的是,本节中所指IVD在我国规则和国外规则中有不同含义。我国的医疗器械分类规则涵盖IVD用设备和器具,但不涉及体外诊断试剂的分类(仅有相关表述“第七条 体外诊断试剂按照有关规定进行分类”)。而下面提到的国外的医疗器械分类规则中,IVD用的设备、器具和试剂均不包括在内。
2.1 我国非IVD医疗器械分类规则
我国新版的《分类规则》[4]于2015年7月14日以国家食品药品监督管理总局令第15号的形式发布,该文件对有源、无源、侵入、植入、作用时间等影响医疗器械风险的因素进行了表述和定义,并以附表的形式对各种情形下产品的管理类别进行了限定。
在规则附表中,根据不同的结构特征和是否接触人体,将医疗器械划分为:无源接触人体器械、无源非接触人体器械、有源接触人体器械、有源非接触人体器械。附表对无源接触人体器械,在不同作用时间(暂时使用、短期使用、长期使用),不同侵入程度(皮肤/腔道(口)、创伤/组织、血循环/中枢)的情况下的管理类别进行了确定;对有源接触人体器械,按照失控后可能造成的损伤程度(轻微损伤、中度损伤、严重损伤)划分管理类别;对非接触人体器械,则按照对医疗效果的影响程度(基本不影响、轻微影响、重要影响)划分管理类别。
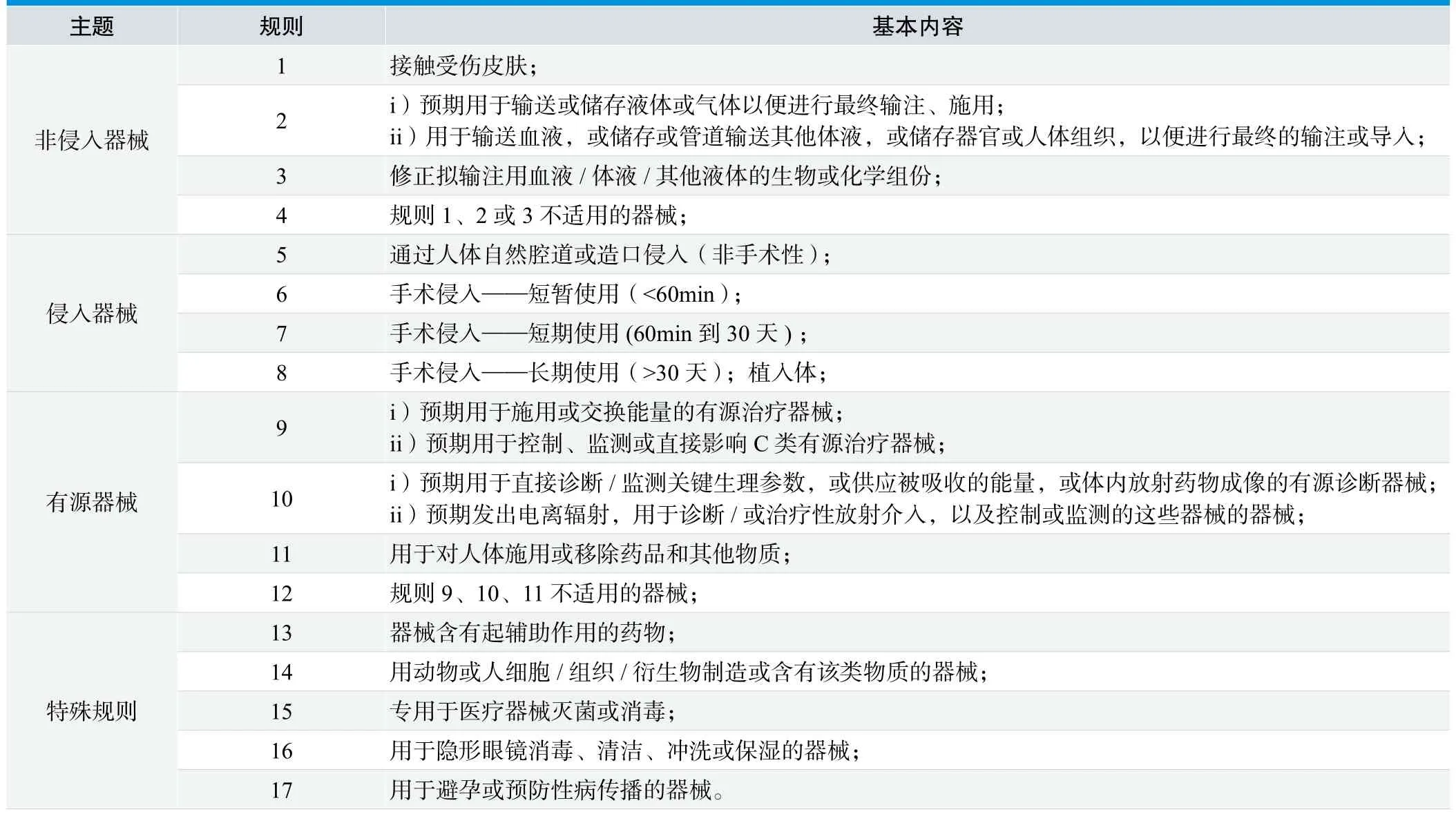
表1. GHTF分类规则基本框架
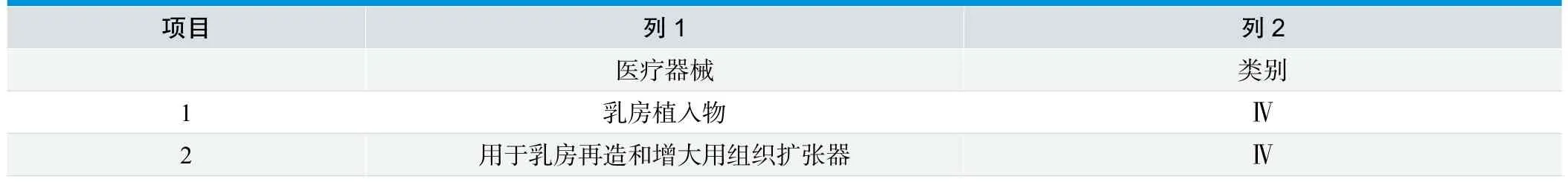
表2.加拿大非IVD医疗器械分类规则特例
在规则附表的基础上,《分类规则》在正文第六条中罗列了十二条原则,分别对包类产品、附件、监控医疗器械的产品、药械组合、可吸收产品、敷料、无菌提供的器械、矫形器械、计量测试功能器械、内窥镜下器械等特殊情况进行了进一步补充说明。
2.2 国外非IVD医疗器械分类规则简介
笔者整理欧盟、美国、日本、加拿大、新加坡、GHTF的相关文件发现,美国没有分类规则一类的文件,其他几国则在不同程度上采用了GHTF的规则[15](见表1),其中:日本采用了较老的版本,以15条规则对非IVD医疗器械进行了分类(隐形眼镜用液体、避孕器械未作为规则列出);新加坡则采用的GHTF最新版本的分类规则,仅根据需要做了细节修改。欧盟现行的指令在采用GHTF规则时仅做了细节修改,并将血袋类产品的分类单独作为一条规则列出。欧盟2012年通过的法规提案[9]中,在原有的规则8和规则9中,加入了有源植入医疗器械按最高管理类别Ⅲ类管理的表述。此外,法规新增了3条规则(共21条),将纳米材料制造或含有纳米材料的医疗器械、用于自体血成分分离回输的器械等器械的管理类别分为Ⅲ类。
加拿大的规则[14]在基本构架上与GHTF类似,主要分为侵入、非侵入、有源、特殊规则几类情况,但具体表达方式和内容有所不同,较大的差异为隐形眼镜用液体、避孕器械未作为规则列出,而加拿大医疗器械(非IVD)分类规则根据需要新增了两条特殊规则,分别为:
规则15:任何一种材料类医疗器械,出于模具或模型的架构和布置,卖给健康护理专业人士或药剂师,以满足个体需要时,按照最终完成的医疗器械类别进行分类。
规则16:规则1-15之外,本规则见表2,列1的医疗器械按照列2进行分类。
3.国内外体外诊断医疗器械分类规则对比
2013年,我国食药监总局依据《医疗器械监督管理条例》、《体外诊断试剂注册管理办法(试行)》(国食药监械[2007] 229号)等有关规定,制定发布了《6840 体外诊断试剂分类子目录》[17]。目录中明确了各个种类体外诊断试剂的预期用途和管理类别(分为第一类、第二类、第三类),共涉及产品品种766个。2014年发布的《体外诊断试剂注册管理办法》[18](国家食品药品监督管理总局令第5号)中第十七条,根据产品风险程度由低到高,以类似清单的形式将各种体外诊断试剂分为第一类、第二类、第三类产品。
2008年,GHTF发布了IVD医疗器械分类原则[16],以7条分类原则将IVD医疗器械(含设备、试剂)分为A、B、C、D四类。到目前为止,欧盟是以98/79/EC中Annex II清单的形式指导不同风险产品的监管,未对其进行管理分类。在欧盟理事会新的法规提案中,对该体系进行调整,采用了GHTF的规则。在新加坡、加拿大等国,采用了与GHTF相同或类似的IVD分类规则。
日本的分类规则中,对于分析仪器和试剂分
别进行了概念性阐述,根据体外诊断试剂则根据诊断信息风险,进行等级分类,分为Ⅰ、Ⅱ、Ⅲ共三个等级,并结合监管实际情况,制定了关于IVD的JMDN数据库。
4.思路分析
我国新版分类规则[4]在某些方面借鉴了国外的分类思路,例如参考欧盟、GHTF指南等[8,15]有关分类的情况,细化了“侵入器械”、“植入器械”的内容,增加了“皮肤”、“腔道(口)”、“创
伤”、“组织”、“血液循环系统”、“中枢神经系统”、“具有计量测试功能的医疗器械”、“慢性创面”等用语的说明,新增“独立软件”的概念等。
这些定义或概念有的与GHTF/欧盟保持了一致,如参考GHTF最新文件,植入器械的定义里面包含了可吸收的情况(这一思路在欧盟文件中尚未体现),参考欧盟的Ⅰ-测量、Ⅰ-无菌类产品的管控措施(欧盟这些产品管控措施与Ⅱa类一致),明确无菌形式提供、具有计量测试功能的医疗器械管理类别不低于第二类。
另一方面,结合我国监管实际和规则的整体逻辑性,我国的规则中一些概念与国际情况有所不同,例如我国侵入器械的概念(强调以手术方式侵入)和GHTF手术侵入器械的概念更接近,而且为了方便管理分类的确定,将较低风险的非无菌、重复使用等情况排除,另作表述。此外,我国关于不同作用时间的划定也与GHTF/欧盟等不同,主要的差异在于“短时使用”这一概念的划分时间点,我国为24h,GHTF/欧盟等为60min。
除了参考国外相关法规文件的概念和思路之外,我国分类规则同时体现了一些我国监管实践总结和特有政策,如第六条中明确了关于医用敷料、矫形器械的管理类别,附表中的“计划生育器械”等。
体外诊断医疗器械方面,在《体外诊断试剂注册管理办法》[18]的正文中,对我国体外诊断分类体系进行了规定。由于该分类体系载于法规正文中,因而比分类规则或目录具有更强的稳定性,更为权威,但同时由于法规制修订周期长,导致该分类体系难以根据新技术涌现、监管形势变化做出即时的调整,其法规滞后性更为明显,对于新产品、新技术的容纳性较差。
目前,GHTF、加拿大、新加坡等都采用分类规则对IVD类器械进行分类,欧盟也拟由清单形式转为分类规则,这些分类规则或作为单独的文件,或作为法规的附件,均没有写入法规正文;另一方面,我国非IVD医疗器械采用的分类规则和分类目录均为独立的技术文件,笔者建议我国体外诊断试剂可以考虑将分类规则作为一个独立的技术法规文件进行制修订,与其独立的分类目录配合使用。
5.结语
随着医疗器械市场的国际化,我国与国外监管方的交流也逐步增多,沟通机制日益成熟。2013年,经国务院批准,我国正式加入国际医疗器械监管机构论坛(IMDRF)。我国医疗器械分类管理改革工作中,充分参考分析了国外相关的管理思路,反复讨论了这些思路在我国社会环境和行业现状下的适用性,最终形成了立足我国国情,参照国际思路,适应行业需求的管理思路。新版《医疗器械分类规则》的制修订充分体现了这一工作思路。
随着我国医疗器械市场进出口贸易的增长,国际医疗器械监管方的交流将越来越重要,互相之间的影响将日趋明显。目前,我国正在开展和深化医疗器械分类管理改革工作,国际上成熟的分类管理思路,特别是那些在较大范围内已取得公认的监管措施,可作为工作中重要的参考资料,为相关工作的开展提供指引和借鉴。
[1] 国务院办公厅. 医疗器械监督管理条例[Z].2014-03-07.
[2] 国家药品监督管理局. 医疗器械分类目录[Z].2002-08-28.
[3] 国家食品药品监督管理总局.第一类医疗器械产品目录[Z]. 2014-05-30.
[4] 国家食品药品监督管理总局.医疗器械分类规则[Z]. 2015-07-14.
[5] The council of the European communities. COUNCIL DIRECTIVE of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices[Z]. 2007-09-05.
[6] The council of the European communities. COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 concerning medical devices[Z]. 2007-09-05.
[7] The European parliament and the council of the European Union. DIRECTIVE 98/79/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 27 October 1998 on in vitro diagnostic medical devices[Z]. 2011-12-20.
[8] European Commission. Medical devices: Guidance document - Classification of medical devices[Z]. 2010-06.
[9] European Commission. Proposal for a REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL on medical devices, and amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009[Z]. 2012-09-26.
[10] European Commission. Proposal for a REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL on in vitro diagnostic medical devices[Z]. 2012-09-26.
[11] United States Congress. Federal Food, Drug, and Cosmetic Act[Z]. 2014 (Amendment).
[12] U.S. Food and Drug Administration. Code of Federal Regulations Title 21—Food And Drugs Chapter I—Food And Drug Administration Department Of Health And Human Services Subchapter H—Medical Devices[Z]. 2015-04-01 (update).
[13] 日本厚生劳动省医药食品局. 关于修改高度管理医疗器械、管理医疗器械以及一般医疗器械的等级分类规则[Z]. 2013-05-10.
[14] Health Canada. Medical Devices Regulations(SOR/98-282,Amended)[Z]. 2011-12-16.
[15] The Global Harmonization Task Force. Principles of Medical Devices Classification[Z]. 2012-11-02.
[16] The Global Harmonization Task Force. Principles of IVD Medical Devices Classification [Z].2008-02-19.
[17] 国家食品药品监督管理总局. 6840 体外诊断试剂分类子目录(2013版)[Z]. 2013-11-26.
[18] 国家食品药品监督管理总局. 体外诊断试剂注册管理办法[Z]. 2014-07-30.
Comparison and Analysis of Rules and Management Concepts Aboutmedical Device Classification in China and World-wide
ZHOU Liang-bin1WU Yi-ming1LI Wei-song1ZHANG Chun-qing2
1 Guangdong Medical Device Quality Surveillance and Test Institute (Guangzhou 510663)
2 National Institutes For Food and Drug Control (Beijing 100050)
New version of China “Classification rules of medical devices” has been released in July 2015, officially implemented since January 2016. In order to facilitatefurtherunderstanding and mastering oftheclassification rules, relevant laws and regulations ofthe EU, the United States, Japan and other countries were summarized, collated and analyzed, the national classification management concepts were analyzed, similarities and differences of classification rules between countries/region were compared.
medical device, classification, rule, management concept
1006-6586(2016)04-0026-06
R194
A
2016-01-12
猜你喜欢
现代仪器与医疗(2022年1期)2022-04-19
河北环境工程学院学报(2021年1期)2021-03-19
生物医学工程学进展(2021年3期)2021-01-20
少儿画王(3-6岁)(2020年4期)2020-09-13
质量安全与检验检测(2019年3期)2019-07-31
支部建设(2019年36期)2019-02-20
质量安全与检验检测(2018年6期)2018-12-28
新教育时代·教师版(2016年27期)2016-12-06
经济师(2016年10期)2016-12-03
西夏学(2016年2期)2016-10-26
