
固体酸催化丁烯-异丁烷烷基化反应机理层面动力学研究
——反应网络生成
2016-12-14 06:21刘晓宇郭锦标王鑫磊李永祥慕旭宏
石油学报(石油加工) 2016年6期
刘晓宇, 郭锦标, 王鑫磊, 李永祥, 慕旭宏,周 祥
(中国石化 石油化工科学研究院, 北京 100083)
固体酸催化丁烯-异丁烷烷基化反应机理层面动力学研究
——反应网络生成
刘晓宇, 郭锦标, 王鑫磊, 李永祥, 慕旭宏,周 祥
(中国石化 石油化工科学研究院, 北京 100083)
深入研究固体酸催化丁烯-异丁烷烷基化反应的机理,构建了以生成主要产物为导向的机理层面反应网络。反应网络由一系列动力学和热力学平衡控制的化学反应组成。根据热力学稳定性和产物选择性,反应体系中结合分子水平和集总水平的反应组分划分尺度,对反应组分进行管理,共包含107个反应组分,实现了与实验数据的匹配。在不损失动力学信息的前提下,通过比较各基元反应的反应速率,对反应类型加以限定,制定了不同类型基元反应的反应规则,反应网络共包含441个基元反应,有效提高了模型求解效率,并利用实验数据验证了反应网络的准确性。
固体酸催化烷基化; 机理层面; 动力学模型; 反应网络; 反应规则
研发分子筛作为固体酸烷基化催化剂的技术难点在于改善其快速失活和选择性差的问题[1]。普遍认为,引起失活的原因是聚合生成的不饱和高聚物在催化剂活性中心上发生不可逆吸附或堵塞孔道[2],因此分子筛烷基化催化剂研发时要侧重于大孔径分子筛的改性[3]。通过实验手段,从分子筛构型和改性的角度,考察孔径大小、硅/铝比、酸性中心分布、酸强度等因素对产物分布和催化剂活性的影响,进而认识催化剂物性与反应物转化的规律性[4-6]。
为进一步明确反应物转化规律,研究者对固体酸催化丁烯异丁烷烷基化反应体系进行了不同尺度的动力学研究。Nayak等[7]针对烷基化反应中的6个关键反应,建立了路径层面动力学模型,证明分子内负氢转移反应和聚合反应是影响催化剂活性的主要反应。Sanchez-Castillo等[8]在机理层面考察了各类型基元反应对反应物中异丁烷转化率的影响,通过灵敏度分析,得到其中31个基元反应具有主要动力学信息。龙军等[9]利用分子模拟方法研究了丁烯-异丁烷烷基化反应体系,通过考察各类基元反应的活化能,搭建了由反应物到异辛烷产物的反应网络。Martins等[10-11]在机理层面建立单事件动力学模型,反应体系中包括反应中间体正碳离子,对可能发生的所有类型基元反应进行详实描述,并利用布尔邻接矩阵生成反应网络,包含3130个基元反应和753个反应组分,选用14个动力学参数,成功地预测了不同碳数产物(C5~C8)的分子组成。但其反应网络中包含非主要动力学信息的反应,导致模型规模庞大,求解困难,还容易引起模型预测结果中包含热力学不稳定的中间产物。
对于固体酸催化烷基化体系所发生的表面反应,活性中心的浓度和酸强度与反应物转化规律存在定量关系,并且反应中间体的结构与产物组成有关。因此,建立机理层面动力学模型,对产物组成及收率实现分子水平的预测,反映动力学参数与原料性质、操作变量、催化剂之间存在的内在联系,可以更好地为新型催化剂研发、反应器设计和工艺流程优化提供理论支持。笔者将对固体酸催化丁烯-异丁烷烷基化反应体系进行细致研究,基于热力学稳定性和产物选择性,从对反应组分管理和基元反应类型限定两方面设计反应网络,以预测C5~C9主要产物组成为导向,构建涵盖主要动力学信息的反应网络,在机理层面研究固体酸催化烷基化反应体系。
1 实验部分
1.1 反应条件
采用等温固定床反应器评价催化剂催化烷基化反应活性。以异丁烷(濮阳市中炜精细化工有限公司产品)和丁烯(中国石化燕山石化公司产品)混合物为原料,其中异丁烷含量远大于丁烯含量,选用某Y分子筛为催化剂。反应趋于稳定后,分别在1.5、3.5、5.5、7.5、9.5、11.5 h时,采用安捷伦公司7890A气相色谱仪在线分析烷基化产物组成。
1.2 实验结果
固体酸催化丁烯和异丁烷烷基化反应的主要产物为C5~C9烷烃,烯烃含量(w<0.3%)相对较少,主要产物的名称列于表1。气相色谱在线分析可以对产物中的C5~C8组分达到分子水平的划分,但无法识别C9~C12的具体组成。烷烃产物中无直链烷烃,目标产物二甲基己烷(DMH)和三甲基戊烷(TMP)为主要组分,其中DMH有2,4-DMH、2,3-DMH、3,4-DMH、2,5-DMH 4种同分异构体,TMP有2,2,4-TMP、2,3,4-TMP、2,3,3-TMP3种同分异构体,而无2,2,3-TMP,因为2,2,3-TMP和2,3,3-TMP的色谱峰重叠。副产物中除异戊烷外,C6、C7中以带有二甲基的产物居多。

表1 固体酸催化丁烯和异丁烷烷基化反应主要产物
2 反应机理分析
烷基化反应遵循正碳离子反应机理,由进料中的烯烃接受B酸中心提供的质子,生成正碳离子,然后发生一系列反应,如图1所示。在固体酸催化烯烃烷基化体系中,反应中间体为吸附态正碳离子。Kazansky等[12]利用AB从头算法结合13C魔角旋转核磁共振推断出,在固体酸催化烯烃烷基化体系中,正碳离子与B酸中心生成C—O键,以能量较低的化学吸附形式存在。Janik等[13]和Comma等[14]针对固体酸催化烯烃烷基化体系,利用分子模拟手段证实了上述观点。因此,在机理层面动力学模型中,将反应中间体设定为吸附态正碳离子,并且叔(Ter)、仲(Sec)、伯(Pri)正碳离子的稳定性依次降低。
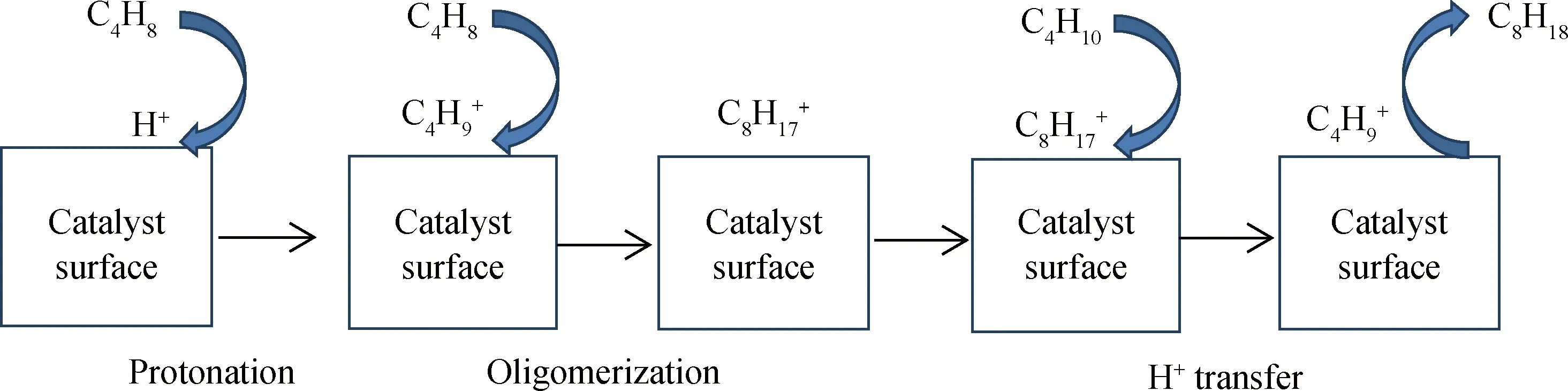
图1 固体酸催化丁烯与异丁烷烷基化的反应过程
固体酸催化烯烃烷基化反应体系包括8种基元反应类型[10],分别为质子化反应、去质子化反应、PCP(Protonated cyclopropane)异构化反应、分子内氢转移反应、分子内甲基转移反应、聚合反应、β-断裂反应和分子间负氢转移反应。
3 反应网络构建
机理层面反应网络由数目庞大的动力学和热力学平衡控制的基元反应组成。在Martins等[10-11]所建立的固体酸催化烯烃烷基化单事件模型的预测结果中包含31个烷烃(C30~C90)和103个烯烃(C3=~C9=),而本实验数据中,产物烷烃和烯烃种类均少于该单事件模型的预测结果。根据热力学稳定性,部分反应中间体和中间产物不易生成或快速转化为其他组分。因此,基于热力学稳定性和产物选择性,在不损失动力学信息的前提下,笔者从反应组分管理和基元反应类型限定两方面着手,构建以生成主要产物为导向的机理层面反应网络。
3.1 反应组分管理
根据产物分布,在C3~C9范围内,选取质量分数大于0.3%的组分作为模型的预测对象,并根据热力学稳定性选取必要中间产物和反应中间体。对于体系中的烷烃限定规则包括,①对于碳数R小于或等于8(R≤8)的正碳离子,区分不同支链数和支链位置;②对于9≤R≤12的正碳离子,不区分支链数和支链位置。对于体系中的烯烃限定规则包括,①不区分双键位置;②只区分不同支链数和支链位置。对于体系中的正碳离子限定规则包括,①区分仲/叔正碳离子;②区分带有正电荷碳原子位置;③区分不同支链数和支链位置;④不区分R>9的正碳离子的支链位置。按照上述反应组分限定规则,模型中包含21个烷烃、22个烯烃、64个正碳离子,共107个组分。其中对于R>9的正碳离子,根据其吸附态生成焓的分布,按照其正碳离子结构、氢质子位置、支链数进行归并,共划分为21个正碳离子集总。
3.2 反应网络构建
根据各基元反应速率和热力学稳定性,对反应体系中各基元反应类型作出限定依据,即①根据产物分布几乎无R>9的烯烃,设定R>9的烯烃不发生去质子化反应;②根据热力学稳定性,设定体系中无伯正碳离子和甲基生成;③非高温情况下,异丁烷无法发生脱氢和脱甲基反应[10];④分子内氢/甲基转移反应为热力学控制的快速平衡反应,趋于生成热力学稳定的正碳离子。反应活化能Esec→ter、Esec→sec、Eter→ter、Eter→sec依次增加[10];⑤叔正碳离子发生分子间负氢转移反应的反应速率常数远大于仲正碳离子发生分子间负氢转移反应的反应速率常数,同时仲正碳离子异构化为叔正碳离子的反应速率常数远大于其发生分子间负氢转移反应的反应速率常数[15],而且由于体系中异丁烷含量远大于其他烷烃,因此有根据认为分子间负氢转移反应主要发生在异丁烷与叔正碳离子之间;⑥模型中只考虑直链正碳离子与一甲基正碳离子之间的PCP异构化反应[16]。因此,在以上限定的基础上,根据反应机理,相对应制定反应规则。
(1) 质子化反应的规则:设定为可逆反应,去质子化反应为其逆反应,对于R<10的烯烃均可发生质子化反应对应生成R<10的正碳离子。
(2) PCP异构化反应的规则:设定为可逆反应,限定发生在直链正碳离子与一甲基的正碳离子之间,反应体系中R>3的直链正碳离子均可发生PCP异构化反应生成具有带有一甲基的正碳离子。
(3) 分子内氢/甲基转移反应的规则:均设定为无直链变化的可逆反应,限定发生在R≤12的正碳离子之间。CR-sec+↔CR-ter+、CR-sec+↔CR-sec+、CR-ter+↔CR-ter+、CR-ter+↔CR-sec+4种类型的分子内氢转移反应和分子内甲基转移反应均可发生。
(4) 聚合反应的规则:设定为由正碳离子进攻烯烃双键中具有较少取代基的碳原子,生成大分子正碳离子。限定反应体系中3≤R≤9的正碳离子与3≤R≤9的烯烃可以发生聚合反应,生成R≤12的大分子正碳离子。
(5)β-裂化反应的规则:设定为正碳离子中带有正电荷的碳原子的α位和β位之间发生断键,生成小分子的正碳离子和烯烃。限定体系中碳数范围在6≤R≤12的正碳离子均可发生β-裂化反应,生成3≤R≤9的正碳离子和3≤R≤9的烯烃。
(6) 分子间氢转移反应的规则:设定为发生在异丁烷和叔正碳离子之间。限定体系中碳数范围R≤12的叔正碳离子均可与异丁烷发生分子间氢转移反应,对应生成碳数范围为3≤R≤12的烷烃和叔丁基正碳离子。
根据上述反应规则,考察各基元反应的反应速率和主要产物分布,选取包含主要动力学信息的反应类型,生成反应网络,如图2所示。其中分子内氢/甲基转移为无支链变化的异构化反应,烷基化反应的目标产物TMP主要有2,2,4-TMP、2,3,4-TMP、2,3,3-TMP、2,2,3-TMP等同分异构体;DMH主要有2,4-DMH、2,3-DMH、3,4-DMH、2,5-DMH等同分异构体,各同分异构体之间经过一系列分子内氢/甲基转移反应进行相互转化,导致各同分异构体产物选择性不同。分子内氢/甲基转移反应是反应体系中反应数目最大的基元反应类型,因此笔者选取了具有主要动力学信息的TMH+和DMH+分子内氢/甲基转移反应,搭建了由反应物(丁烯和异丁烷)转化为主要烷基化产物(C5~C9)的机理层面反应网络,共包含441个基元反应。TMH+和DMH+分子内氢/甲基转移反应如图3和图4所示,反应网络中基元反应的数目列于表2第1 列(Reaction network 1)。

图2 固体酸催化丁烯异丁烷烷基化反应网络
3.3 反应网络验证
将所建的固体酸催化丁烯异丁烷烷基化反应的机理层面动力学模型反应网络应用于Martins等[10-11]所述的反应体系,验证模型反应网络的准确性,结果示于图5。从图5可以看出,机理层面动力学模型对固体酸催化丁烯异丁烷烷基化反应主要产物C5~C8组分的预测值与实验值吻合较好,相关因子在0.9以上,可见所建机理层面动力学模型反应网络符合了反应物分子→反应中间体/中间产物→产物的反应历程,可以准确预测产物组成。
相对于Martins[10-11]所述单事件反应网络,本反应网络中基元反应由3130个减少至441个(见表2),显著提高了模型求解效率。由于所建模型中对反应组分管理结合了分子水平和集总水平的反应组分划分尺度,因此在C8以上组分所涉及的基元数目有效降低的同时,并不降低模型对主要产物预测结果的准确性;并且通过比较基元反应的反应速率,对反应体系中热力学不稳定的中间产物和反应中间体参与的基元反应进行限定的同时,并不损失动力学信息。
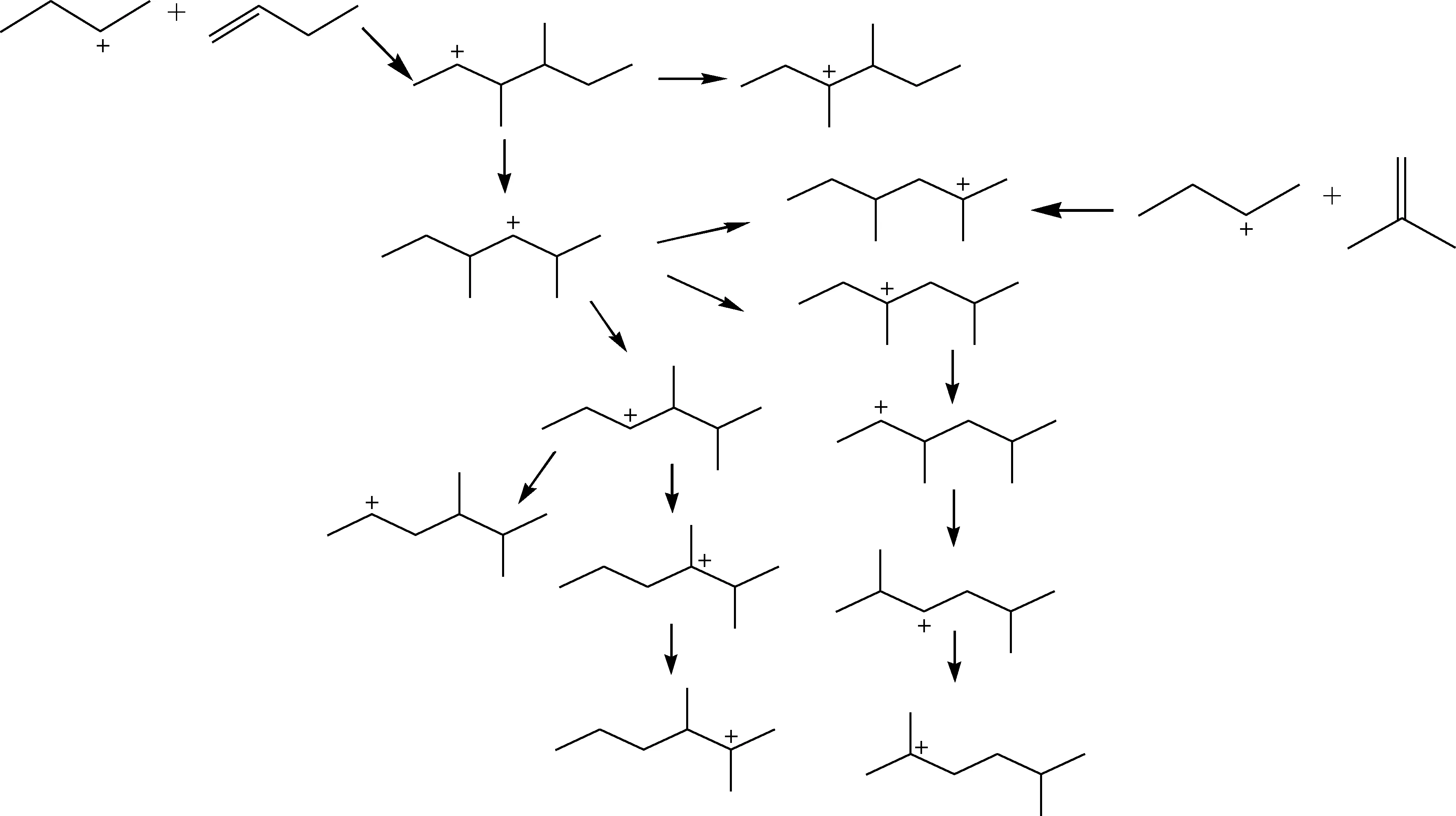
图3 DMH+分子内氢/甲基转移反应

图4 TMP+分子内氢/甲基转移反应

表2 固体酸催化丁烯异丁烷烷基化反应网络基元反应数目
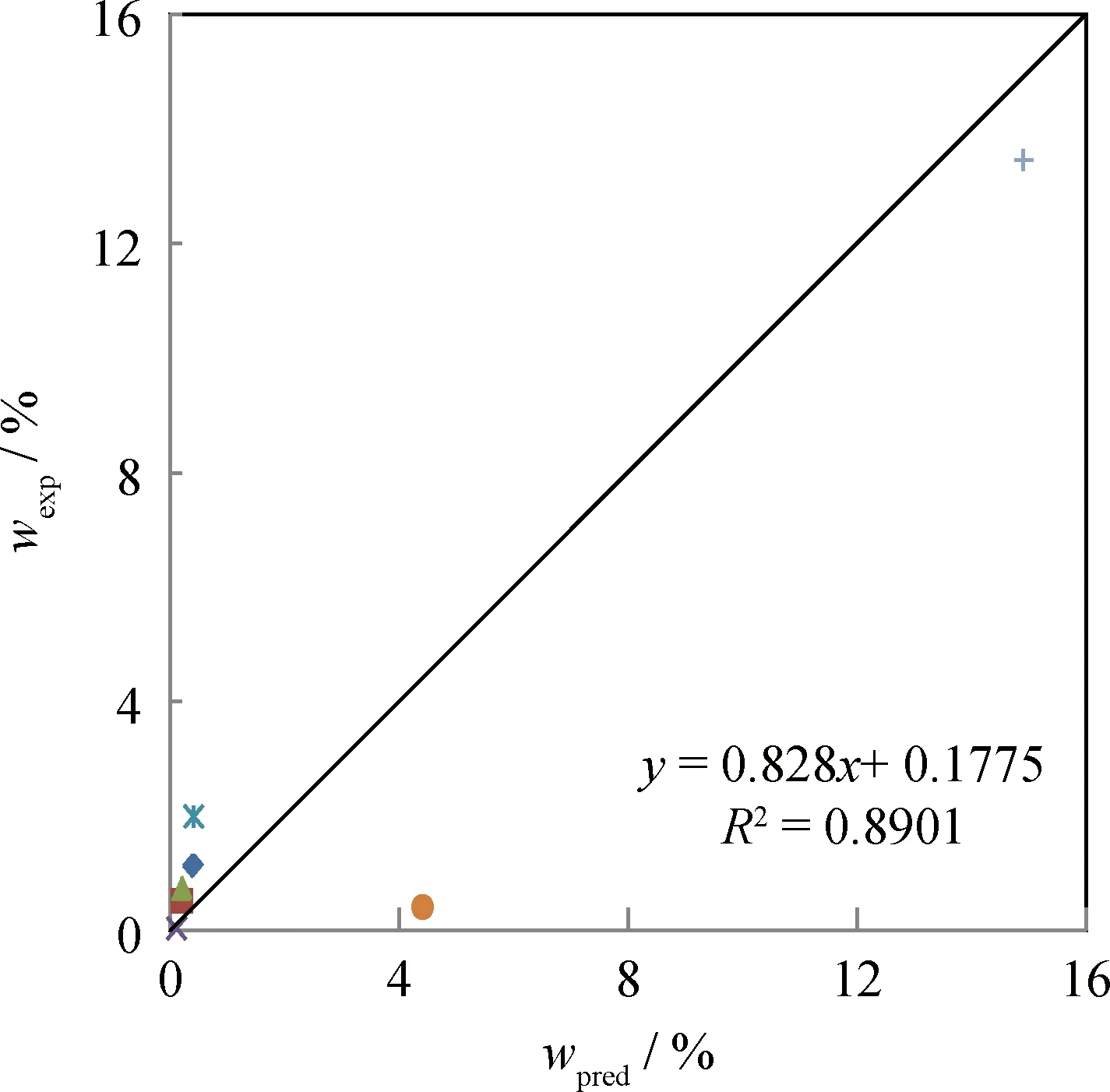
图5 固体酸催化丁烯异丁烷烷基化反应主要产物的模型预测值与实验值的对比
4 结 论
(1) 根据正碳离子反应机理,生成由基元反应组成的固体酸催化丁烯异丁烷烷基化反应网络。一方面依据产物选择性和热力学稳定性,对反应组分进行管理;另一方面由于实验数据中无C8以上组分的具体分子组成,结合了分子水平和集总水平的反应组分划分尺度,共划分为107个反应组分。分子水平组分包含39个分子(C5~C8)和43个正碳离子(C5~C9),集总水平组分包含4个分子集总(C9~C12)和21个正碳离子集总(C10~C12)。因此,反应组分共包含21个烷烃组分、22个烯烃组分,相对应按照正碳离子结构、氢质子位置、支链数对R>9正碳离子进行归并,划分为64个正碳离子组分。
(2) 根据产物分布,对质子化反应加以限定;根据热力学稳定性,对β-断裂反应加以限定;通过比较基元反应的反应速率,对PCP异构化反应和分子间负氢转移反应加以限定,以此对各基元反应制定了相应的反应规则。以生成主要产物为导向对分子内氢转移和分子内甲基转移反应进行考察,选取了涵盖主要动力学信息的基元反应,搭建了固体酸烷基化机理层面反应网络。该反应网络共包含441个基元反应,保留了关键动力学信息,明确了反应物的转化规律和C5~C9主要产物的生成路径,可以有效地提高模型求解效率。模型预测结果与实验数据吻合度较好,相关因子在0.9以上。
[1] WEITKAMP J, TRAA Y. Isobutane/butene alkylation on solid catalysts where do we stand?[J].Catalysis Today, 1999, 49: 193-199.
[2] BARTHOLOMEW C H. Mechanisms of catalyst deactivation[J].Applied Catalysis A: General, 2001, 212: 17-60.
[3] SIMPSON M F, WEI J, SUNDARESAN S. Kinetic analysis of isobutane/butene alkylation over ultrastable H-Yzeolite[J].Ind Eng Chem Res, 1996, 35: 3861-3873.
[4] YOOK, SMIRNIOTIS P G. The influence of Si/Al ratios of synthesized beta zeolites for the alkylation of isobutane with 2-butene[J].Applied Catalysis A: General, 2002, 227: 171-179.
[5] FELLER A, ZUAZO I, GUZMAN A. Common mechanistic aspects of liquid and solid acid catalyzed alkylation of isobutane withn-butene[J].Journal of Catalysis, 2003, 216: 313-323.
[6] SEKINE Y, ICHIKAWA Y, TAJIMA Y. Alkylation of isobutane by 1-butene over H-betazeolite in CSTR: Part 1 Effects of zeolite-structures and synthesis methods on alkylation performance[J].Journal of the Japan Petroleum Institute, 2012, 55(5): 299-307.
[7] NAYAK S V, RAMACHANDRAN P A, DUDUKOVIC M P. Modeling of key reaction pathways: Zeolite catalyzed alkylation processes[J].Chemical Engineering Science, 2010, 65: 335-342.
[8] SANCHEZ-CASTILLO M A, AGARWAL N, MILLERET C. Reaction kinetics study and analysis of reaction schemes for isobutane conversion over USY zeolite[J].Journal of Catalysis, 2002, 205: 67-85.
[9] 龙军, 何奕工, 代振宇. 异丁烷-丁烯烷基化体系基元反应网络研究[J].石油学报(石油加工), 2010, 26(1): 1-7.(LONG Jun, HE Yigong, DAI Zhenyu. Elucidation of elementary reaction network for isobutane-butene alkylation system[J].Acta Petrolei Sinica (Petroleum Processing Section), 2010, 26(1): 1-7.)
[10] MARTINIS J M, FROMENT G F. Alkylation on solid acids: Part 1 Experimental investigation of catalyst deactivation[J].Ind Eng Chem Res, 2006, 45: 940-953.
[11] MARTINIS J M, FROMENT G F. Alkylation on solid acids: Part 2 Single-event kinetic modeling[J].Ind Eng Chem Res, 2006, 45: 954-967.
[12] KAZANSKY V B, FRASHM V, van SANTEN R A. Quantum-chemical study of hydride transfer in catalytic transformation of paraffins on zeolites[J].Studies in Surface Science and Catalysis, 1997, 105: 2283-2290.
[13] JANIK M J, DAVIS R J, NEUROCK M. A density functional theory study of the alkylation of isobutane with butene over phosphotungstic acid[J].Journal of Catalysis, 2006, 224: 65-77.
[14] BORONAT M, VIRUELA P M. CORMA V A. Reaction intermediates in acid catalysis by zeolites: Prediction of therelative tendency to form alkoxides or carbocations as a function of hydrocarbon nature and active site structure[J].J Am Chem Soc, 2004, 126: 3300-3309.
[15] GUILLAUME D. Network generation of oligomerization reactions: Principles[J].Ind Eng Chem Res, 2006, 45: 4554-4557.
[16] WATSON B A, KLEIN M T, HARDING R H. Mechanistic modeling ofn-heptane cracking on HZSM-5[J].Ind Eng Chem Res, 1996, 35(5): 1506-1516.
Kinetic Analysis of Butene Alkylationon With Isobutane onSolid Acids——Generation of Reaction Network
LIU Xiaoyu, GUO Jinbiao, WANG Xinlei, LI Yongxiang, MU Xuhong, ZHOU Xiang
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
The reaction mechanism of solid acid-catalyzed alkylation of butane with isobutane was studied. The reaction network considering the key product composition was generated at mechanistic level, which consisted of a series of reactions controlled by kinetics and thermodynamic constraints. Based on the thermodynamic stability and product selectivity, the reaction network contained 107 reactions to be managed on molecule and lump level and compared with experimental results. Reaction rules of different elementary steps were formulated via defining reaction types, based on comparison of reaction rates without loss of kinetic information. The established reaction network, whose accuracy was verified by experimental results, contained 441 elementary steps, and had impressive effectiveness on the solution of the model.
solid acid-catalyzed alkylation; mechanistic level; kinetic model; reaction network; reaction rule
2016-02-17
刘晓宇,女,博士研究生,从事烷基化反应动力学方面研究
周祥,男,高级工程师,博士,从事计算机在石油化工生产的应用研究;E-mail: zhoux.ripp@sinopec.com
1001-8719(2016)06-1205-07
TE624.4
A
10.3969/j.issn.1001-8719.2016.06.017
猜你喜欢
计算机工程与应用(2023年1期)2023-01-13
炼油与化工(2021年3期)2021-07-06
化工管理(2020年19期)2020-07-28
科学导报(2018年30期)2018-05-14
石油石化绿色低碳(2018年5期)2018-03-20
石油炼制与化工(2016年6期)2016-04-06
合成化学(2015年2期)2016-01-17
化学工业与工程(2015年1期)2015-02-10
无机化学学报(2014年1期)2014-02-28
