
皮肤淋巴组织增生性病变的临床病理分析*
2016-12-06 03:05:51李芬章培张文燕陈琼涂媛石海鹏何林李钒刘卫平
西部医学 2016年11期
李芬 章培 张文燕 陈琼 涂媛 石海鹏 何林 李钒 刘卫平
(1.成都市第二人民医院病理科, 四川 成都 610017; 2.四川大学华西医院病理科, 四川 成都 610041)
·论著·
皮肤淋巴组织增生性病变的临床病理分析*
李芬1章培1张文燕2陈琼1涂媛1石海鹏1何林1李钒1刘卫平2
(1.成都市第二人民医院病理科, 四川 成都 610017; 2.四川大学华西医院病理科, 四川 成都 610041)
目的 探讨皮肤淋巴组织增生性病变的临床病理特征,以期提高病理诊断质量,规范临床治疗。方法 复习16例皮肤淋巴组织增生性病变患者临床病理资料,分析常见皮肤淋巴组织增生性病变的基本构成、大体皮损及组织学形态、免疫表型特点,探讨病理诊断和诊断要点。结果 16例患者男性11例,女性5例,男女比2.2∶1,中位年龄52.5岁。 病程2月~16年不等,临床皮损形态多样且无特异性。病理诊断为非肿瘤性的皮肤淋巴细胞浸润3例,淋巴瘤13例包括蕈样霉菌病4例、淋巴瘤样丘疹病3例、原发皮肤间变性大细胞淋巴瘤2例、ALK阳性间变性大细胞淋巴瘤累及皮肤1例、皮肤NK/T细胞淋巴瘤1例,弥漫大B细胞淋巴瘤2例。10例行EBER1/2 原位杂交检测,仅1例皮肤NK/T细胞淋巴瘤阳性。9例行TCRγ基因或IgH/IgK受体基因重排分析,蕈样霉菌病1例、间变性大细胞淋巴瘤2例、 淋巴瘤样丘疹病1例和弥漫大B细胞淋巴瘤1例检出克隆性重排。结论 本组皮肤淋巴组织增生病变中,T细胞淋巴瘤居多,以蕈样霉菌病最为常见;与EB病毒关系不密切;诊断与鉴别诊断需密切结合临床。
淋巴组织增生; 淋巴瘤; 皮肤; 临床病理分析
皮肤淋巴组织增生性病变可以原发于皮肤,也可为系统性淋巴组织肿瘤的继发累及;病理改变可以为反应性淋巴组织增生、淋巴瘤瘤前病变,也可以是不同生物学行为的淋巴瘤。由于病例数量少,皮肤淋巴组织增生性病变的病理诊断甚为困难。随着世界卫生组织(WHO)和欧洲癌症研究与治疗小组(EORTC)原发皮肤淋巴瘤分类(2005年)的提出,皮肤科临床医师和病理医师采用同一种诊断标准,既不断提高病理诊断质量,也更加规范临床治疗[1]。本研究总结了16例皮肤淋巴组织增生性病变的临床病理特征,旨在积累临床病理诊断经验。
1 资料和方法
1.1 病例来源 收集自2014年6月至2016年6月成都市第二人民医院皮肤活检标本7094例,筛选出淋巴组织增生性病变疑似病例20例,经两名病理医生复习诊断,明确诊断为皮肤淋巴组织增生性病变16例。
1.2 方法
1.2.1 临床特征观察 收集患者现病史、既往史、皮损大体形态、主要诊断经过等资料,并电话随访。
1.2.2 形态学观察 所有病例的活检组织均经10%中性缓冲福尔马林溶液固定,石蜡包埋,3 μm切片,苏木素-伊红染色。总结病变模式、肿瘤细胞形态特点及伴随改变。
1.2.3 免疫表型分析 采用Envision法,DAB显色。选用的一抗包括CD20、 CD79a、MUM-1、BCL-6、CD10、BCL-2、CD5、CD23、CD43、CD21、CD30、CD56、CD138、ALK-1、Ki-67、Kappa、lambda、GranzymeB、TIA-1、CD2、CD4、CD8、CD7(福州迈新生物技术有限公司),CD3ε、PCK、EMA(广州安必平医药科技有限公司)。
1.2.4 EBER1/2原位杂交 地高辛标记的针对EBV编码的小分子mRNA(EBER1/2)的肽核酸探针(福州泰普)在石蜡切片上进行原位杂交,桥接HRP标记的抗Dig-DAB显色系统。以鼻咽癌组为阳性对照。细胞核呈现棕黄色颗粒信号即为阳性。
1.2.5 基因重排检测 石蜡包埋组织快速DNA提取方法提取DNA,DNA扩增采用BIOMED-2系统引物,PCR反应在MJ Research PTC200型PCR仪上进行,产物采用聚丙烯酰胺凝胶电泳、异源双链核酸分子分析技术或毛细管电泳结合基因扫描(ABI3500基因分析仪)进行分析。详细操作按照科室基因检测实验常规进行。
1.2.6 诊断标准 参照淋巴造血系统肿瘤WHO分类(2008),其中原发皮肤淋巴瘤按照1WHO-EORTC分类确定。
2 结果
2.1 临床特征 16例患者中男性11例,女性5例,男女比2.2∶1.0。年龄22岁~86岁,平均年龄54.9岁,中位年龄52.5岁。病史2月~16年不等,其中6例多个部位两次皮肤活检,10例为一次活检。
2.2 皮肤淋巴组织增生性病变临床资料 16例患者临床资料见表1。病理诊断结果显示:3例(18.8%)为皮肤淋巴细胞浸润;余13例为肿瘤性病变(83.3%)。肿瘤性病变中成熟T/NK细胞肿瘤11例(11/13,84.3%),包括蕈样霉菌病(Mycosis Fungiodes,MF)4例,淋巴瘤样丘疹病3例, ALK阳性间变性大细胞淋巴瘤累及皮肤1例,原发皮肤间变性大细胞淋巴瘤2例以及结外NK/T细胞淋巴瘤(鼻型)1例, 余2例(2/13,15.4%)为弥漫大B 细胞肿瘤。16例患者临床皮损及病理诊断,见表1。

表1 16例皮肤淋巴组织增生性病变临床资料
2.3 大体特点 4例蕈样霉菌病患者均有明显皮肤瘙痒症状。3例泛发全身,1例局限于头颈部、胸部皮肤。临床皮损多样。经典型斑片期表现为红斑鳞屑性、苔藓样、大疱样,大疱样皮损疱壁破裂后伴有浅表糜烂,斑块期表现为高于皮面的形态不规则浸润性斑块(图1A),肿瘤期为结节性肿块。其中两例患者同时出现多个时期病变。1例表现为全身皮肤弥漫发红,呈现红皮病改变;2例亲毛囊型MF一例表现为躯干红斑鳞屑性斑片、头顶部秃发性斑块(图1B),1例表现为头颈部及胸部播散性丘疹、结节。1例伴有淋巴结肿大。3例淋巴瘤样丘疹病皮损出现于躯干、四肢,均有反复出现的大小不一丘疹、红斑症状,皮损可自行消退但反复发作,皮肤消退后可留下瘢痕,其中1例出现溃疡性病变且2月不消退(图1C)。皮肤原发间变性大细胞淋巴瘤2例:分别表现为大腿聚集性皮肤结节和躯干红斑丘疹伴逐渐扩大的溃疡。1例继发性间变性大细胞淋巴瘤则表现为大腿皮肤溃疡、腹股沟淋巴结肿大。皮肤NK/T细胞淋巴瘤1例:表现为小腿皮肤多个溃疡、浸润性红斑(图1D)。3例皮肤淋巴细胞浸润表现面部、颈部皮肤红斑、丘疹。

图1 典型病例表现
Figure 1 Typical cases
注:A.病例1, 蕈样霉菌病: 四肢浸润性斑块;图B.病例3,亲毛囊性蕈样霉菌病:患者头顶部头发秃发性斑块,头皮表面粗糙,红斑鳞屑;C.病例5,淋巴瘤样丘疹病:躯干、四肢皮肤多发性红斑、丘疹,溃疡,皮损新旧不一,陈旧性病变遗留瘢痕,新发皮损溃疡伴少许淡黄色分泌物;D.病例13,皮肤NK/T细胞淋巴瘤(鼻型):小腿皮肤浸润性红斑、溃疡、边界不清
2.4 组织学改变
2.4.1 蕈样霉菌病 4例均为两个或两个以上部位活检,各个部位具有不同的病变模式:银屑病样、苔藓样、药疹样、湿疹样、大疱样,均具有基底细胞液化变性等界面皮炎改变。经典型病变有亲表皮现象明显,可见Pautrier微脓肿,亲毛囊型肿瘤细胞浸润破坏毛囊,2例均可见毛囊粘蛋白沉积,但界面皮炎样改变及淋巴细胞亲表皮现象不如经典型明显。肿瘤细胞在真皮内呈散在或聚集性分布,呈halo细胞样或核呈脑回样;伴淋巴细胞、中性粒细胞浸润。4例均有嗜酸性粒细胞浸润。
2.4.2 皮肤原发间变性大细胞淋巴瘤 1例表面溃疡;1例肿瘤细胞在真皮层及皮下脂肪结节状浸润。溃疡底部或真皮结节内大小不一的异型淋巴样细胞弥漫性浸润,胞浆淡染或嗜碱,核偏位或居中,核仁明显,可见多核瘤巨细胞或R-S样细胞,伴淋巴细胞及中性粒细胞浸润。2例均伴有嗜酸性粒细胞浸润。
2.4.3 ALK阳性间变性大细胞淋巴瘤累及皮肤 表面溃疡。形态同4.2的肿瘤细胞弥漫浸润真皮及皮下脂肪组织,部分肿瘤细胞呈梭形。3例均伴有嗜酸性粒细胞浸润。
2.4.4 淋巴瘤样丘疹病 2例表皮完整,病变局限于真皮内,未浸润皮下脂肪。小淋巴样细胞为主的淋巴细胞分布于真皮血管周围,可见散在或小灶性聚集性分布的大细胞,大细胞胞浆淡染或嗜碱,可见R-S样细胞。另1例皮肤溃疡者,异型大细胞聚集性分布,形态学难以与间变性大细胞淋巴瘤区分。3例均伴有嗜酸性粒细胞浸润。
2.4.5 皮肤NK/T细胞淋巴瘤 表皮溃疡。中等大小的异型淋巴样细胞弥漫分布,从溃疡底部累及皮下脂肪,伴坏死。可见血管中心性浸润,未见嗜酸性粒细胞浸润。
2.2.6 弥漫大B细胞淋巴瘤 表皮完整。真皮及皮下脂肪组织内见大量大的中心母细胞或免疫母细胞样异型细胞弥漫浸润,可见散在分布的瘤巨细胞。凋亡易见,未见中性粒细胞浸润。
2.4.7 皮肤淋巴细胞浸润 表皮完整。无淋巴细胞亲表皮现象,部分病例表皮下见无细胞浸润带,小淋巴细胞结节状分布,有淋巴滤泡,浆细胞浸润,散在胞浆淡染的大细胞散在分布,其中未见嗜酸性粒细胞浸润。
2.5 免疫表型、原位杂交、基因重排检测结果
2.5.1 蕈样霉菌病 肿瘤性淋巴细胞呈CD3(+)、CD7(-)、CD4(+)、CD8(-)、CD2(-)、CD5(-)、CD20(-)、CD30(-)EMA(-)、CD56(-)、ALK-1(-)、CD117(-)、MPO(-)、PCK(-)、S-100(-)、其中1例肿瘤细胞出现大细胞转化:TIA-1(部分+)、GranzymeB(部分+)。1例TCRγ基因重排阳性。
2.5.2 原发皮肤间变性大细胞淋巴瘤 CD3(+)、CD20(-)、CD30(+,弥漫成片)EMA(-)、CD56(-)、ALK-1(-)、TIA-1(+)、GranzymeB(部分细胞+)。其中2例TCRγ基因重排阳性。
2.5.3 ALK阳性间变性大细胞淋巴瘤累及皮肤 CD3(+)、CD20(-)、CD30(+,弥漫成片)、ALK-1(+)、EMA(部分+),TIA-1(+)、GranzymeB(部分+)。
2.5.4 淋巴瘤样丘疹病 CD20(-)、 CD3(+)、CD30(+,阳性细胞散在或小簇状分布)(图2A)、EMA(-)、CD56(-)、TIA-1(+)、GranzymeB(部分细胞+)。1例TCRγ基因重排阳性,2例TCRγ基因重排阴性。
2.5.5 皮肤NK/T细胞淋巴瘤(鼻型) CD20(-)、 CD3(+)、CD5(-)、CD56(+)、TIA-1(+)、GranzymeB(细胞+)。
2.5.6 弥漫大B细胞淋巴瘤 CD20(+)、CD3(-)、BCL-6(+)、CD10(+)、MUM-1(-)。
2.5.7 皮肤淋巴细胞浸润显示混合性T、 B淋巴细胞浸润,散在分布的大细胞为活化的CD30阳性的淋巴细胞(图2B)。CD21显示淋巴滤泡有完整滤泡树突状细胞网。其中1例TCRγ基因重排阴性,一例IGH/IGK受体基因重排阴性。
2.5.8 EBER1/2原位杂交 10例进行了检测,1例皮肤NK/T细胞淋巴瘤为阳性,余9例(包括MF2例、淋巴瘤样丘疹病1例、原发皮肤间变性大细胞淋巴瘤1例、ALK阳性间变性大细胞淋巴瘤累及皮肤1例、弥漫大B细胞淋巴瘤2例、皮肤淋巴细胞浸润2例)均为阴性。
2.6 随访 随访时间1月~2年。2例MF患者死亡:1例诊断后一月因突发心室纤颤死亡,1例MF 全身淋巴结肿大、放弃治疗后3月死亡。余均带病生存。
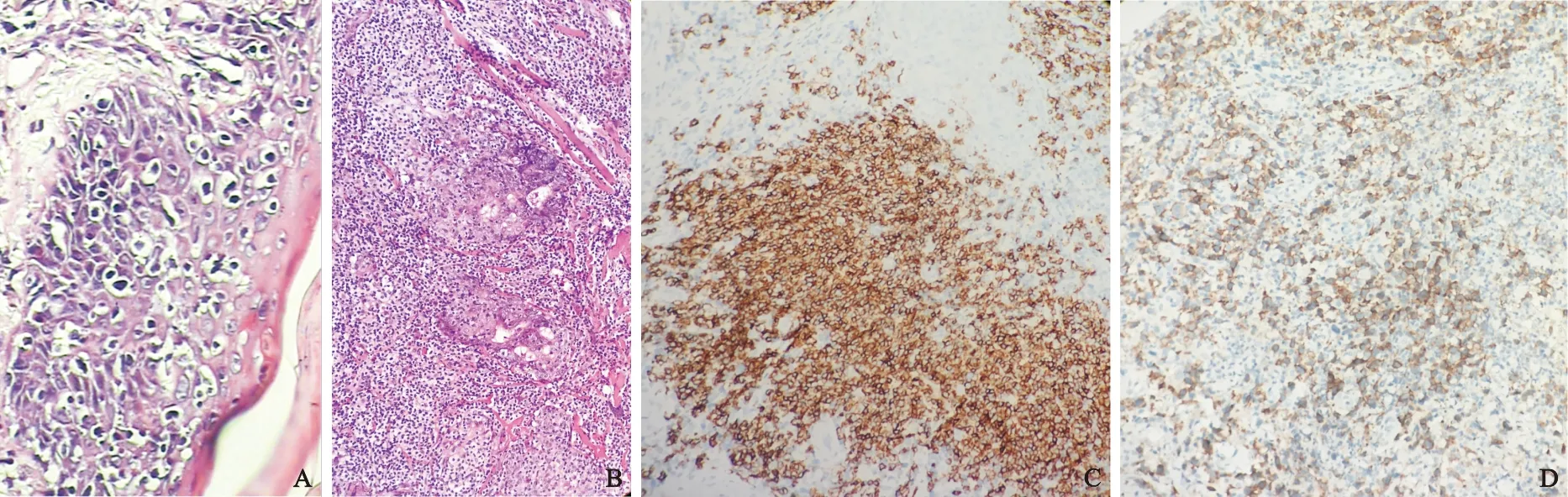
图2 皮肤淋巴组织增生性病变的形态图
Figure 2 Cutaneous lymphoid hyperplasia
注:A.病例2, 蕈样霉菌病: 肿瘤细胞亲表皮,表皮内Pautrier微脓疡 HE 高倍放大;B.病例3,蕈样霉菌病:肿瘤细胞亲毛囊,毛囊结构部分破坏 HE 中倍放大;C.病例16,皮肤淋巴组织增生:EnVision法 中倍放大,真皮内浸润淋巴细胞CD20阳性,并呈结节状分布;D.病例5,淋巴瘤样丘疹病 EnVision法 中倍放大CD30阳性细胞呈散在或小簇状分布。
3 讨论
本组皮肤淋巴组织增生性疾病患者年龄跨度大(22~86岁),最年轻者病变为系统性ALK阳性间变性大细胞淋巴瘤累及皮肤。男性明显多于女性,其中4例淋巴瘤样丘疹病均为男性,具有性别优势。病程长短不一,其中MF组病史最长,诊断前病程2~16年不等。临床表现多样,缺乏特异性,活检前确诊率极低,除两例疑诊淋巴瘤外,余者考虑湿疹、结核、扁平苔藓、孢子丝菌病、血管炎、脂膜炎、多形红斑、慢性溃疡、包块性质待诊等。
本组病例大多数为肿瘤性病变(13/16,81.3%),且以T细胞淋巴瘤占绝对优势(12例),与文献报道一致[2]。其病理诊断尤以MF及淋巴瘤样丘疹病最为困难。二者在低倍镜下形态不易与炎症性病变、湿疹样、苔藓样病变等鉴别,常表现为临床皮损表现与镜下形态的严重程度不相符合,因此对高倍镜下细胞形态的辨别、是否为肿瘤性病变以及免疫组化识别细胞来源等要求颇高。值得一提的是,本组10例T细胞淋巴瘤均伴有数量不等的嗜酸性粒细胞浸润,甚至掩盖肿瘤细胞;而3例皮肤淋巴组织增生以混合性淋巴细胞浸润为特点,B细胞数量相对多,可伴浆细胞分化,无嗜酸性粒细胞浸润(大B细胞淋巴瘤亦如此),提示嗜酸性粒细胞的有无可指导免疫标记的选择。
在鉴别诊断方面,MF是最常见的嗜表皮的原发皮肤T细胞淋巴瘤,早期淋巴细胞浸润表皮,产生扁平且伴有少量鳞屑的皮损(斑片期),最终淋巴细胞增生对表皮失去依赖,获得在真皮内和内脏器官增殖能力,形成皮肤斑块和结节(斑块期、肿瘤期),表皮内可以完全没有淋巴细胞浸润,缺乏Pautrier微脓肿[3-5]。故不同时期的MF需要和多种炎症性皮肤病变鉴别。扁平苔藓、玫瑰糠疹、急性痘疮样苔藓样糠疹等炎性皮肤病可以出现T细胞受体基因克隆性重排,此时称为克隆性皮炎[6-11],但临床呈现良性的过程,因此基因重排的结果一定要结合临床综合判读。此外,大细胞转化的MF(Transformation of mycosis fungoides,TMF)肿瘤细胞形态酷似间变性大细胞淋巴瘤,23%(34/148)TMF 可CD30+[12],二者则无法鉴别,必须结合临床病史、患者年龄、皮损形态及临床结局综合分析。
淋巴瘤样丘疹病和原发性皮肤间变性大细胞淋巴瘤同属原发性皮肤CD30阳性T细胞淋巴增殖性疾病,CD30阳性在前者常为真皮内呈小簇状或片状阳性模式,而后者呈弥漫性、累及皮下脂肪的阳性模式[13]。二者在WHO2008分类中ICD编码分别为1和3,性质有别,但鉴别有时非常困难。人们试图运用免疫组化标记如TRAF1, MUM1, BCL2 及CD15区别淋巴瘤样丘疹病与间变性大细胞淋巴瘤、霍奇金等相关淋巴瘤,但结果让人失望[14]。2008版淋巴瘤样丘疹病分A、B、C三型[15],其中B型特点为具有脑回样核的异性小细胞嗜表皮浸润,需要与MF鉴别,但非常少见(<10%)。A型和C型的主要不同在于背景炎细胞的多少和肿瘤细胞的多少。2016年WHO分类中,又增添了D型、E型及6p25重排的淋巴瘤样丘疹病三型,D型组织学上类似于原发皮肤的侵袭性嗜上皮性CD8+细胞毒性T细胞淋巴瘤,E型组织学上也类似于侵袭性淋巴瘤,但常可自愈。虽然伴6p25重排淋巴瘤样丘疹病累及的基因位点也为DUSP22-IRF4,但组织学与侵袭性原发性皮肤间变性大细胞淋巴瘤相似,但却并不发生播散性皮肤病变或皮肤外病变。2016年分类强调这三个亚型的临床鉴别非常重要[16]。此外,有报道60%的淋巴瘤样丘疹病检测到T细胞受体基因克隆性重排[17],因此必须结合临床病史谨慎解读基因重排结果以资甄别。Cordel等[18]的研究表明:伴有克隆性T细胞重排和年龄大是淋巴瘤样丘疹病进展为淋巴瘤的高危险因素, 需要临床给予更严格的管理和随访。
由于原发皮肤间变性大细胞淋巴瘤未检出(2;5)(p23;q35)异位[19],因此ALK阳性更支持系统性间变性大细胞淋巴瘤的皮肤累及而不是原发皮肤淋巴瘤。Lee 等[20]对比研究原发皮肤的间变性大细胞淋巴瘤与继发性皮肤ALK阳性间变性大细胞淋巴瘤后发现:两组病变有相同的组织学形态、免疫表型,继发性肿瘤皮肤病变比原发性肿瘤病灶大;原发肿瘤比继发性肿瘤预后更好,但发生于大腿的原发性肿瘤的预后相对大腿外的原发性肿瘤预后差;原发性肿瘤皮损的严重程度对是否进展和复发有提示作用。
对于皮肤NK/T细胞淋巴瘤和弥漫大B细胞淋巴瘤,由于有相对特异的免疫表型及细胞特点,相对容易,但后者病理诊断中一定要注意提醒临床进行全面的身体检查以排除系统性淋巴瘤累及皮肤的可能。
4 结论
皮肤淋巴增生性疾病患者男性居多,年龄跨度大,临床缺乏特异性。肿瘤性病变为主,多为T细胞淋巴瘤,尤以MF最为常见。MF、淋巴瘤样丘疹病与皮肤原发间变性大细胞淋巴瘤的鉴别需要密切结合临床,皮肤淋巴组织增生性病变中T细胞受体基因克隆性重排的结果要结合临床、HE形态、免疫组化结果综合分析。除NK/T细胞淋巴瘤外,未见皮肤淋巴组织增生性病变与EB病毒感染相关。
[1]刘卫平, 李甘地.WHO-EORTC关于皮肤淋巴瘤分类简介[J].中华病理学杂志,2007,36(2):133-135.
[2]陈刚, 李小秋.血液病理学[M].北京:北京科学技术出版社,2013:348-349.
[3]周小鸽, 陈辉树.造血与淋巴组织肿瘤WHO分类[M].第4版.北京:诊断病理学杂志社,2011:535-539.
[4]Song SX, Willemze R,Swerdlow SH,etal. Mycosis fungoides: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop[J]. Am J Clin Pathol, 2013,139(4):466-490.
[5]Massone C, Kodama K, Kerl H,etal. Histopathologic features of early (patch) lesions of mycosis fungoides: a morphologic study on 745 biopsy specimens from 427 patients[J]. Am J Surg Pathol, 2005,29(4):550-560.
[6]Pimpinelli N,Olsen EA, Santucci M,etal. Defining early mycosis fungoides[J]. J Am Acad Dermatol, 2005,53(6):1053-1063.
[7]Wood G,Haettner A,Dummer R,etal.Molecular biology technique for the diagnosis of cutaneous T-cell lymphoma[J].Dermatol Clin,1994,12:231-241.
[8]Wood G,Tung RM,Haeffner AC,etal.Detection of clonal T-cell receptor gamma gene rearrangements in early mycosis fungoides/Sezary syndrome by polymerase chain reaction and denaturing gradient ge l electrophoresis(PCR/GGE) [J]. J Invest Dermatol,1994,103(1):34-41.[9]Dommann SN, Dommann-Scherrer CC,Dours-Zimmermaan MT,etal.Clonal disease in extracutaneous compartments in cutaneous T-cell lymphomas.A comparative study between cutaneous T-cell lymphomas and pseudo lymphomas[J].Arch Dermatol Res,1996,288(4):163-167.
[10] Muche JM,Lukowsky A,Ahnhudt C,etal.Peripheral blood T cell clonality in mycosis fungoides-an independent prognostic marker? [J].Blood,1999,94(4):1409-1417.
[11] Holm N, Flaiq MJ,Yazdi AS,etal.The value of molecular analysis by PCR in the diagnosis of cutaneous lymphocytic infiltrates[J]. J Cutan Pathol, 2002,29(8):447-452.
[12] Jullié ML, Carlotti M, Vivot A Jr,etal.CD20 antigen may be expressed by reactive or lymphomatous cells of transformed mycosis fungoides: diagnostic and prognostic impact[J]. Am J Surg Pathol, 2013,37(12):1845-1854.
[13] 王琳, 刘卫平.皮肤淋巴瘤图解指南[M].第3版.北京:人民卫生出版社,2012 :60-78.
[14] Benner MF,Jansen PM,Meijer CJ,etal.Diagnostic and prognostic evaluation of phenotypic markers TRAF1, MUM1, BCL2 and CD15 in cutaneous CD30-positive lymphoproliferative disorders[J]. Br J Dermatol,2009,161(1):121-127.
[15] 周小鸽, 陈辉树.造血与淋巴组织肿瘤WHO分类[M]. 第四版.北京:诊断病理学杂志社,2011:542-545.
[16] Swerdlow SH, Campo E,Pleri SA,etal. The 2016 revisiom of the World Health Organization classification of lymphoid neoplasms[J]. Blood,2016,127(20): 2375-2390.
[17] Kadin ME,Pathobiology of CD30+ cutaneous T-cell lymphomas[J]. J Cutan Pathol,2006,33(Suppl 1):10-17.
[18] Cordel N,Tressières B,D'Incan M,etal.Frequency and Risk Factors for Associated Lymphomas in Patients With Lymphomatoid Papulosis[J]. Oncologist, 2016,21(1):76-83.
[19] DeCoteau JF, Butmarc JR,Kinney MC,etal. The t(2;5) chromosomal translocation is not a common feature of primary cutaneous CD30+ lymphoproliferative disorders: comparison with anaplastic large-cell lymphoma of nodal origin[J]. Blood,1996,87(8):3437-3441.
[20] Lee WJ, Moon IJ, Lee SH,etal. Cutaneous anaplastic large-cell lymphoma (ALCL): A comparative clinical feature and survival outcome analysis of 52 cases according to primary tumor site[J]. J Am Acad Dermatol, 2016,74(6):1135-1143.
Cutaneous lymphoproliferative disorders: a clinicopathologic analysis
LI Fen1, ZHANG Pei1,ZHANG Wenyan2,et al
(1.DepartmentofPathology,ChengduSecondPeople'sHospital,Chengdu610017,China;2.DepartmentofPathology,WestChinaHospital,SichuanUniversity,Chengdu610041,China)
Objective To investigate the clinicpathologic features of cutaneous lymphoproliferative disorder. Methods 16 cases of cutaneous lymphoproliferative disorders were collected and analyzed. Constitution, skin eruptions, histopathologic feature, immunophenotype, Epstein-Barr virus status and clinical course were studied. Results Among the 16 patients, the ratio of male to female was 2.2: 1. Median age was 52.5 years old. Clinical courses of disease ranged from 2 years to 16 years. Various and non-specific skin eruptions were observed. 16 case were diagnosed as lymphocytic infilitration of skin (3 cases), mycosis fungiodes(4 cases), primary cutaneous anaplastic large cell lymphoma(2 cases), ALK positive anaplastic large cell lymphoma infiltration of the skin(1 cases), Lymphomatoid papulosis(3 cases), Extranodal NK/T-cell lymphoma nasal type(1 cases ) and diffuse large B-cell lymphoma(2 cases). Only one positive reaction of EBER1/2 in situ hybridization was detected in Extranodal NK/T-celllymphoma, nasal type. TCRγgene or IgH Ig gene colonel rearrangements were detected in 5/9 cases. Conclusion Lymphomas, especially T- cell lymphomas are the common diseases of cutaneous lymphoproliferative disorders. Mycosis Fungiodes is the most common cutaneous T cell lymphoma. The definite correlation of cutaneous lymphoproliferative disorders with Epstein-Barr virus was not be found.Both pathologic diagnosis and differentiate diagnosis of cutaneous lymphoproliferative disorders are challenging, and should be made with clinical information.
Lymphoproliferative disorders; Lymphoma; Cutaneous;Clinicpathologic analysis
国家自然科学基金(81272626)
张文燕,本刊审稿专家,E-mail:zhangwenyanpath@163.com
R 739.5
A
10.3969/j.issn.1672-3511.2016.11.015
2016-08-15;编辑: 张文秀)
猜你喜欢
现代临床医学(2021年4期)2021-07-31 07:55:34
世界最新医学信息文摘(2021年12期)2021-06-09 08:35:26
中国生殖健康(2020年2期)2021-01-18 02:51:18
中国现代医药杂志(2020年12期)2020-02-06 06:32:30
国际呼吸杂志(2019年3期)2019-03-01 05:39:06
中国生殖健康(2018年2期)2018-11-06 07:10:40
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:05
磁共振成像(2015年1期)2015-12-23 08:52:19
现代医药卫生(2014年18期)2014-03-11 19:33:24
中医研究(2013年5期)2013-03-11 20:26:55
