
乳酸菌基因组研究应用进展
2016-12-02 06:56马长路逄晓阳张书文刘鹭芦晶吕加平
食品与发酵工业 2016年10期
马长路, 逄晓阳, 张书文, 刘鹭, 芦晶,吕加平*
1(中国农业科学院,北京,100193)2(北京农业职业学院,北京,102442)
乳酸菌基因组研究应用进展
马长路1, 2, 逄晓阳1, 张书文1, 刘鹭1, 芦晶1,吕加平1*
1(中国农业科学院,北京,100193)2(北京农业职业学院,北京,102442)
为了更好地了解乳酸菌基因组研究和应用的现状,通过对近几年来国内外乳酸菌基因组相关研究的整理分析,发现目前的3个研究重点:菌种鉴定、菌种改良(基因重组)和基因组学研究。结果表明,对乳酸菌突变体和特异性菌种的基因组研究,结合蛋白组学、代谢组学和相关研究成果,将进一步揭示乳酸菌功能特性的实质,让乳酸菌更好的成为细胞工厂、防腐抑菌和营养保健的家庭成员。
乳酸菌,菌种鉴定,基因重组,基因组学
书面记载发酵食品的生产可以追溯到大约公元前4000年,这可能是“最古老的”生物技术实践。乳酸菌(LAB)一直是在这些应用的核心,从而起到了重要的作用。这个长期的使用传统中,人类利用LAB参与食品发酵,达到理想的效果,同时获得由美国食品和药物管理局认可的安全地位(GRAS)。
乳酸菌是革兰氏阳性微生物,兼性厌氧,主要发酵产物是乳酸。因为在发酵过程中,乳酸菌快速产酸,从而能够防止腐败,延长产品的保质期。此外,乳酸菌还有助于改善发酵的风味、质地和营养价值。
近年来,人们利用基因组相关技术研究了食品,如奶酪、肉、发酵谷物食品、葡萄酒、发酵洋橄榄和食醋中的乳酸菌,也研究了青贮饲料中的乳酸菌,而且还研究了活性物质,如D-乳酸、活性肽、纳豆激酶中的相关的乳酸菌。除了上述应用层面的研究,人们利用基因组相关技术研究了生物模型的构建[1]、代谢工程[2]和易错全基因组扩增技术。本文通过对这些研究的整理与分析,从菌种鉴定、菌种改良(基因重组)和基因组学研究3个方面加以阐述。
1 基因组学在菌种鉴定中的应用
常规乳酸菌菌种的分离鉴定流程是在分离纯化微生物后,通过表型特征、形态学观察,以及生理生化反应分析来实现。随着分子生物学的发展,人们开始从分子和基因水平来研究菌种,PCR技术成为研究的热门。国外对于益生菌菌种的研究主流是以各类乳酸菌菌株的基因指纹特征为基础,建立针对单个乳酸菌的特征性图谱库。表1给出了近年来一些利用基因组学信息开展菌种鉴定的研究成果。
FOLARIN等人在2种传统的非洲发酵谷物食品中,采用微生物基因组分类法,研究土著细菌的基因差异和分布。从尼日利亚北部和南部地区的奥吉和军隅崎中分离获得85个主要的细菌种类。它们的种类通过16S rRNA 基因序列分析结合基于rpoA,pheS和atpA基因和M13-PCR凝胶图谱的多位点序列分析(MLSA)得到确认。结果表明,发酵乳杆菌是从奥吉(71.4%)和军隅崎(84.5%)中分离获得最常见的菌种。其他种类的乳酸菌(LAB)鉴定为植物乳杆菌,Streptococcusgallolyticussubsp.macedonicus和戊糖片球菌。对乳酸菌M13-PCR指纹图谱的非度量多维标度(nMDS)分析表明,同一物种的菌株之间存在克隆多样性[6]。
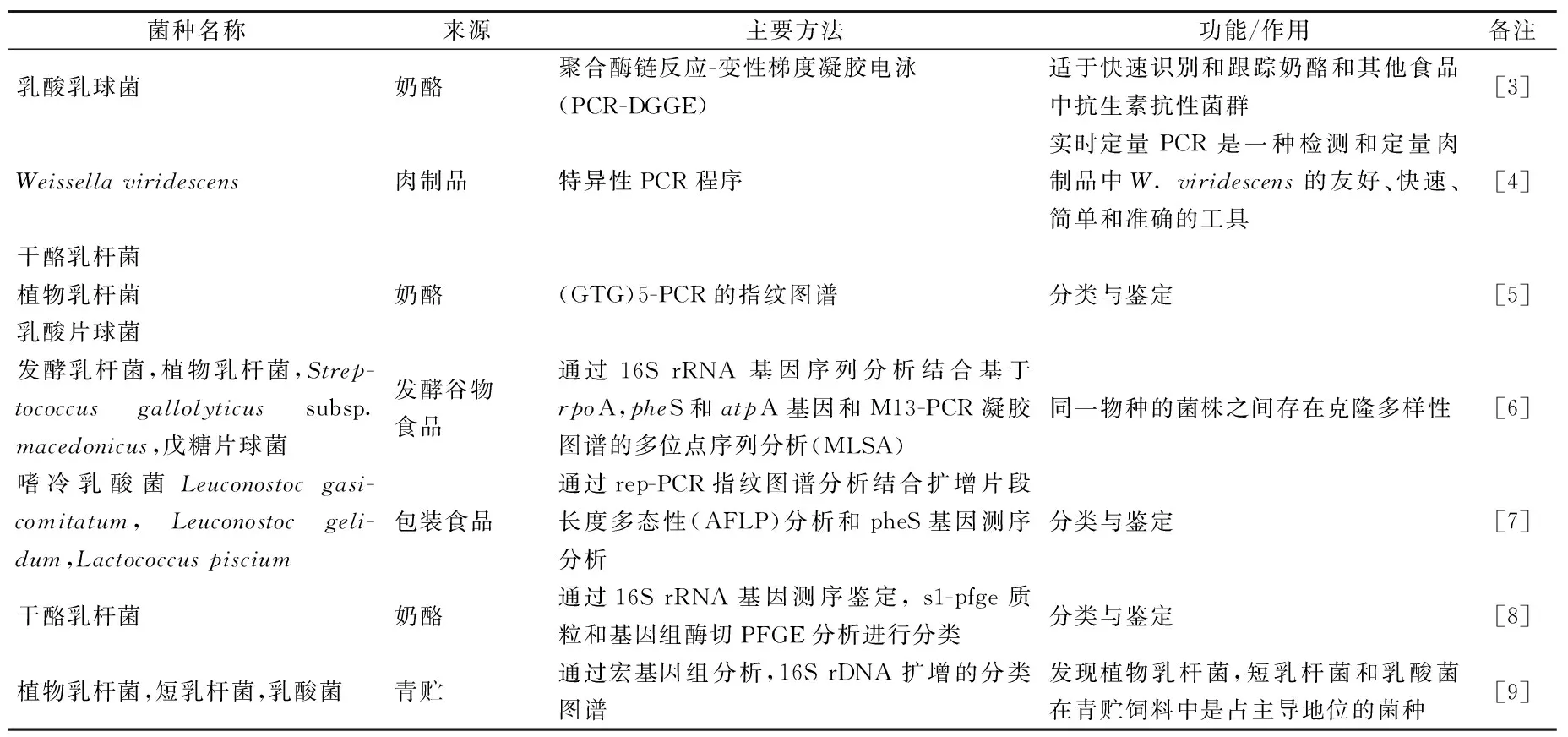
表1 利用基因组信息进行菌种鉴定
VASILEIOS采用多相分类的方法确定了22 ℃, 5天平板培养后,占主导地位的嗜冷乳酸菌的微生物多样性得以恢复。以前,采用时温枚举技术,观测嗜冷菌的乳酸菌(LAB)在储存在低温的33种零售包装食品的污染水平,作为保质期的参考参数,该污染水平有相当大的低估(0.5~3.2 lg CFU/g)。目前研究中,通过rep-PCR指纹图谱分析结合扩增片段长度多态性(AFLP)分析和pheS基因测序分析,共有212株乳酸菌被鉴定[7]。
FELIX等人研究了接种布氏乳杆菌cd034对微生物群落结构的影响。由16S rDNA扩增序列导出的分类群落概况表明,在发酵过程中,乳酸乳球菌的相对丰度减少,同时,属于门变形菌和拟杆菌的细菌的比例在未经处理的青贮发酵过程中有所增加。此外,通过宏基因组分析青贮样品,确认基于16S rDNA扩增的分类图谱。通过筛选宏基因组序列片段,分析读取完整的参考基因组序列,发现植物乳杆菌,短乳杆菌和乳酸菌在青贮饲料中是占主导地位的菌种[9]。
2 基因组学在乳酸菌菌种改良(基因重组)的应用进展
随着生命科学研究领域的不断深入和细化,通过研究乳酸菌的遗传变异及多样性,采用分子生物学手段对乳酸菌进行遗传修饰和改造,以期能够生产某种特定产物,提高某种性能或改善产品品质。表2列出一些利用乳酸菌基因组信息改造菌株的研究数据。
EnantiopurityLactobacilluscoryniformis在30 ℃培养时主要发酵产物为D-乳酸。然而,在发酵温度高于40 ℃时能够检测到一定量的L-乳酸。因为光学纯的乳酸是由手性D-或L-乳酸脱氢酶(D-LDHs或L-LDHs)催化丙酮酸合成的,较高热稳定性的L-LDHs被认为是降低L.coryniformis生产D-乳酸光学纯度的关键因素之一。为了证实这个假设,在Escherichiacoli中合成并且表达了基于L.coryniformis基因组信息的2种D-LDH基因和6种L-LDH基因。结果表明,同D-LDH1相比具有较高的热稳定性的L-LDHs可能是高温发酵时D-乳酸纯度降低的主要原因[10]。
冯浩等研究者构建整合型表达载体pFY008,以HisH基因作为同源重组的靶位点,通过同源重组将含有纳豆激酶原基因的表达盒PnisA-aprN整合到乳酸菌的基因组上,实现纳豆激酶在乳酸乳球菌中表达[14]。
YE等研究戊糖乳杆菌ATCC 8041的耐酸性,是通过使用随机引物和TaqDNA聚合酶对基因组DNA易错扩增而得以改进。仅仅一轮突变后,突变体(MT3)被发现,在pH 3.8的1 L MRS培养基,培养36 h,能够完全消耗20 g/L葡萄糖,产生乳酸,得率为95%,而在相同条件下野生型菌株均未观察到生长或生产乳酸。突变体MT3耐酸性保持稳定遗传至少25代[13]。
郭晶等研究者利用基因组改组方法筛选出促进干酪成熟的非发酵剂乳酸菌。采用紫外线和亚硝基胍两种传统的诱变方法对植物乳杆菌进行诱变,通过检测自溶度、氨肽酶、产酸能力指标获得6株氨肽酶活性和自溶度均有所提高的突变菌株。以获得的突变菌株为出发菌株,对其进行基因组改组,经自溶度、氨肽酶、产酸能力筛选,获得1株遗传性能稳定的菌株,其氨肽酶活力为34.01个酶活单位,比出发菌株提高了3.09倍,自溶度为56.45%,比出发菌株提高了3.16倍[15]。
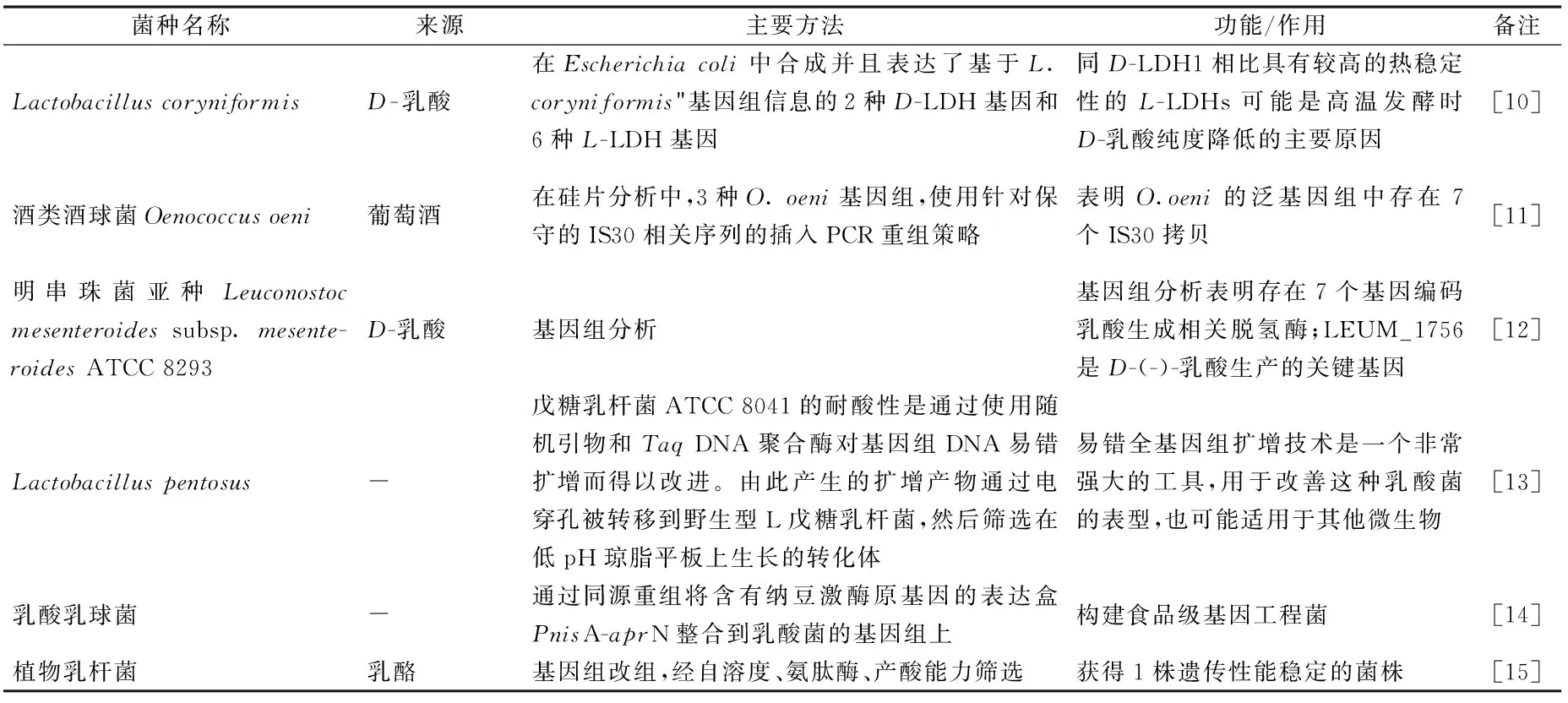
表2 利用基因组信息进行菌种改良(基因重组)
3 基因组学研究进展
基因组学由美国科学家THOMAS R于1945年提出, 是指对基因组进行作图、序列分析、基因定位及功能分析的科学。功能基因组学Functional genomics的研究又被称为后基因组学,是利用结构基因组学提供的信息和产物, 通过在基因组或系统水平上全面分析基因的功能, 使得生物学研究从对单一基因或蛋白质的研究转向对多个基因或蛋白质同时进行系统研究。功能基因组学的研究包括基因功能发现、基因表达分析及突变检测。它采用一些新的技术对成千上万的基因表达进行分析和比较, 力图从基因组整体水平上对基因的活动规律进行阐述。表3列出近些年乳酸菌基因组信息研究的数据。
如今,人们对促进健康的功能性食品的兴趣与日俱浓,从而使消费者持有更高的期望,超过基本的营养诉求,更加注重健康。 膳食蛋白提供了一个丰富的生物活性肽资源,这是隐藏在天然蛋白质内的一个潜在状态,需要酶促蛋白水解释放。生物活性肽能够在体内消化和/或食品加工过程中产生。乳酸菌是最广泛使用的微生物发酵食品的发酵剂,并通过其蛋白水解系统,促进膳食蛋白质的生物活性肽的释放。在体外和体内研究表明多种生物学功能归结为生物活性肽,如抗菌,免疫调节,增强矿物质的吸收,增强抗血栓形成,抗高血压药物和抗氧化的活性。在这些产品中巨大的复杂性和广泛的相对肽丰度的动态范围,严重挑战现有分析方法的能力。然而,功能和比较基因组学研究,以及蛋白质组学方法提供了丰富的知识,揭示这些乳酸菌利用食物蛋白释放生物活性肽的方式[16]。
布氏乳杆菌属于异型发酵乳酸菌,是青贮微生物组的普通一员。L.布氏cd034全基因组测序是在罗氏基因组测序FLX平台上进行的。它被发现有4个复制子,1个圆形的染色体,和3个质粒。预测环状染色体是编码2 319个蛋白,含有基因组岛和2个原噬菌体。原噬菌体同其余染色体具有显著不同的G+C含量。它具有1个完整的酶磷酸途径酶的所有基因,而缺乏2个必要的糖酵解酶。这证实了cd034 L.布氏的分类为一种专性异型发酵乳酸菌。一组基因被认为参与乳酸降解途径,同时一组基因被证实可能参与植物细胞壁聚合物的分解。此外,一些位于染色体的编码S-层蛋白和两个CRISPR系统的基因,属于子类I-E和II-A。最大的质粒pcd034-3被认为编码57个基因,其中包括一个多糖合成基因簇,而2个小质粒pcd034-1和pcd034-2的功能仍保持神秘。基于保守的标记基因rpoA序列比较的系统发育分析发现,L.布氏cd034和布氏乳杆菌菌株同短乳杆菌和植物乳杆菌菌株相比更为密切相关。同其他全序列测定的和乳杆菌属的成员密切相关的核心基因组比较,L.布氏cd034和最近测序的L.布氏乳杆菌菌株NRRL b-30929存在高度的保守,同时它和L.布氏乳杆菌菌株ATCC 11577和L.brevisssp.ravesensisATCC 27305之间存在更远的关系。L.布氏cd034基因组信息将提供深入研究后基因组的基础,以期优化青贮生产菌株的应用[17]。

表3 利用基因组信息进行组学研究
植物乳杆菌是一类与人类的生活关系密切, 具有多种益生功能的乳酸菌。随着高通量测序技术的广泛应用, 越来越多的植物乳杆菌基因组全序列测定得以实现。以植物乳杆菌基因组为主要研究对象,陈臣等人采用比较基因组学分析和比较了几株测序的植物乳杆菌的基因组特点[18]。
HIKMATE等人从4个西班牙中小企业的自然发酵的洋橄榄中筛选144株乳酸菌,包括乳酸杆菌(81.94%),明串珠菌 (10.42%) 和片球菌(7.64%)。 REP-PCR聚类和phes和rpo基因测序进一步鉴定表明,从不同的中小企业筛选出的乳酸杆菌菌株是戊糖乳杆菌。基因分型结果显示,菌株没有克隆的相关性,而是展现一定的基因组多样性,尤其是乳杆菌和明串珠菌[20]。
传统食醋具有悠久的历史,生产工艺独特,酿造过程中复杂的微生物群落及其代谢产物赋予了传统食醋独特的风味。采用宏基因组学技术对天津独流老醋醋酸发酵过程中细菌群落组成及其多样性进行分析。结果表明:在醋酸发酵前期细菌具有较高的多样性,主成分分析表明与醋酸发酵过程相关的细菌为乳杆菌属(Lactobacillus)、醋杆菌属(Acetobacter)和念珠藻属(Nostoc)。随着醋酸发酵的进行,醋酸菌的含量呈增加趋势,乳酸菌的丰度降低,在整个醋酸发酵过程中乳酸菌的丰度远远高于其他细菌,说明乳酸菌可能对食醋的风味形成具有重要作用[22]。
4 组学与代谢工程
YOSHIDA等人引入植物乳杆菌基因组中2份戊糖乳杆菌xylAB操纵子。由此产生的菌株能够发酵1个25 g/L木糖和75 g/L葡萄糖的混合物,无碳代谢抑制的影响,乳酸产率为0.78 g/g[23]。
VAN等人开展了对于能够适应8%乙醇的植物乳杆菌细胞的全局转录组分析,过表达柠檬酸代谢和细胞包络结构的基因,以及由中央应激调控子HrcA 与CtsR控制的典型应激反应途径中的基因[24]。而且,目前能够获得乙醇耐受型菌株的基因组序列[25]。总之,这些信息肯定会发现适合的基金用于乙醇生产的乳酸菌设计途径。
最近,枯草芽孢杆菌(纳豆)丙氨酸脱氢酶用于乳酸丙氨酸生物合成的潜力被证明。这种酶在42 ℃保留高活性,使其在应用时需要高温显得非常有趣。事实上,这是一个应用于酸奶中的嗜热链球菌的工程策略,典型的生物发酵会产生甜而健康的产品,这个目标是依赖于有机体及其表达系统的有效转型[26]。
最近几年来,通过引入下一代测序培养技术和随之而来测序成本的降低,乳酸菌的基因组项目激增。目前,大约40种相关的工业乳酸菌的基因组都存放在公共数据库,同时报告了100多个基因组测序项目仍在继续(http://www.genome.jp/kegg/; http://www.ncbi.nlm.nih.gov/genome; http://www.jgi.doe.gov/)。利用这些基因组序列促进全基因组芯片的发展,能够分析在任何一个给定的时间的基因表达(转录组分析),能够识别全部基因的基因型-表型联系(比较基因组杂交)[27]。在未来的几年里,测序的进展有望推动转录组数据的数量和质量。
在乳酸菌中通过蛋白质组学开展全蛋白补充说明没有转录组学频繁,主要是由于相关蛋白质组学技术层面的挑战。然而,在应激条件和适应环境变化时乳酸菌蛋白组定量分析已被报道[28-29]。这些研究中的大多数仍然依赖于凝胶为基础的技术,但随着质谱技术的发展,以质谱为基础的蛋白质组学将有更广泛的应用[30]。在体内核磁共振的最大优势是依赖于它的独特能力,提供实时测量的细胞内的终产物或中间产物。特别是生产乳酸的乳酸乳球菌,每当细胞内和细胞外空间的PH值的差异能够充分分离相应的共振时,能够区分细胞内和细胞外的乳酸;这种分离是由于两个部分不同的乳酸质子化程度。从实际角度来看,在葡萄糖谢过程中,当细胞外的PH值低于6时,量化外部和内部的乳酸是可行的[31]。
最近,获得一种改进的乳酸乳球菌的糖酵解反应的动力学模型[32]。随后,这些作者以乳酸乳球菌模型为蓝本,快速开发了化脓性链球菌糖酵解的第一反应动力学模型,从而允许物种间比较。虽然该系统并未确定,呈现一些动力学的非可识别参数,但磷酸变构调节的不同,可能是基于在被链球菌科占据的自然环境的磷酸水平。系统代谢工程的使用将继续开展,构建模型也将更加重要。对于给定的试验条件和数据集,定义和构建最佳的模型方程仍然是一个挑战[33]。
不同类型的不同结构的层级数据的整合也是未来研究的有趣课题[34]。例如,热力学和动力学性质的整合在基因组规模模型的限制,被证明是相关的减少通过消除不可行途径的通量空间解[35]。预计将包含额外的实验数据,如基因表达,蛋白表达,代谢物浓度,和动力学参数,可用于减少基于约束模型的解空间[36]。最近,集成模型代谢和大分子表达模型提出[37],构成一个有前途的和强大的框架,从一个系统的水平和基因组规模定量描述细胞的生理现象,可以预见系统代谢工程乳酸菌将受益于这些方法。
5 展望
对乳酸菌突变体和特异性菌种的基因组研究,结合蛋白组学、代谢组学和相关研究成果,将促进乳酸菌在如下3个方面的研究:细胞工厂,主要指以乳酸菌为载体加工生物活性产物;防腐抑菌,主要指乳酸和其他有机酸的抑菌防腐作用;营养保健,主要包括降血脂、降胆固醇和抗氧化作用。通过各种技术的结合,必将推动乳酸菌基础和应用研究的不断发展。
[1] FILIPE B dos S,WILLEM M de Vos,BAS T.Towards metagenome-scale models for industrial applications — the case of Lactic Acid Bacteria[J].Current Opinion in Biotechnology,2013,24(2):200-206.
[2] PAULA G,ANA L C,SUSANA V,et al.From physiology to systems metabolic engineering for the production of biochemicals by lactic acid bacteria[J].Biotechnology Advances,2013,31(6):764-788.
[3] ANA B F,BALTASAR M.Diversity and dynamics of antibiotic-resistant bacteria in cheese as determined by PCR denaturing gradient gel electrophoresis[J].International Journal of Food Microbiology,2015,214(2):63-69.
[4] ERICA M G, ROMERO-SANTACREU L, JAIME I,et al.A novel real-time PCR assay for the specific identification and quantification ofWeissellaviridescensin blood sausages[J].International Journal of Food Microbiology,2015,215:16-24.
[6] FOLARIN A O, ARJAN N. Molecular characterization of lactic acid bacteria and in situ amylase expression during traditional fermentation of cereal foods[J].Food Microbiology,2012,31(2):254-262.
[7] VASILEIOS P, CINDY S, PAUL De Vos, et al. Psychrotrophic members ofLeuconostocgasicomitatum,LeuconostocgelidumandLactococcuspisciumdominate at the end of shelf-life in packaged and chilled-stored food products in Belgium[J].Food Microbiology,2014,39:61-67.
[8] ANA H F,NOELIA M,ESTHER S L,et al.Lactobacilluscaseistrains isolated from cheese reduce biogenic amine accumulation in an experimental model[J].International Journal of Food Microbiology,2012,157(2):297-304.
[9] FELIX G E, PETRA K, ANDREA P,et al.Metagenome analyses reveal the influence of the inoculantLactobacillusbuchneriCD034 on the microbial community involved in grass ensiling[J].Journal of Biotechnology, 2013, 167(3): 334-343.
[10] Gu S-A, CHANHA J,JEONG C J,et al.Higher thermostability of l-lactate dehydrogenases is a key factor in decreasing the optical purity of d-lactic acid produced fromLactobacilluscoryniformis[J].Enzyme and Microbial Technology,2014,58-59(9):29-35.
[11] FATIMA E G,MARGUERITE D L,ELISABETH B,et al.IS30 elements are mediators of genetic diversity inOenococcusoeni[J]. International Journal of Food Microbiology,2012,158(1) :14-22.
[12] LI L, EOM Hyun-Ju, PARK Jung-Mi,et al.Characterization of the major dehydrogenase related to d-lactic acid synthesis inLeuconostocmesenteroidessubsp.mesenteroidesATCC 8293[J].Enzyme and Microbial Technology,2012,51(5):274-279.
[13] YE Lidan, ZHAO Hua, LI Zhi,et al.Improved acid tolerance ofLactobacilluspentosusby error-prone whole genome amplification[J].Bioresource Technology,2013,135:459-463.
[14] 冯浩,余凤云,单凤娟,等.构建食品级表达纳豆激酶的乳酸乳球菌重组菌株[J].食品科学,2012, 33(21) :208-212.
[15] 郭晶,李晓东,姚春艳,等.基因组改组选育氨肽酶和自溶度高的植物乳杆菌[J].中国乳品工业,2013,41(1):11-14.
[16] SAAVEDRA L, HEBERT E M, MINAHK C, et al. An overview of “omic” analytical methods applied in bioactive peptide studies[J].Food Research International,2013,54(1):925-934.
[17] STEFAN H,DANIEL W,FELIX E,et al.Insights into the completely annotated genome ofLactobacillusbuchneriCD034, a strain isolated from stable grass silage[J].Journal of Biotechnology,2012,161(2): 153-166.
[18] 陈臣,任婧,周方方,等.植物乳杆菌的比较基因组学研究[J].中国生物工程杂志,2013,33(12):35-44.
[19] MORGAN G,MONIQUE Z,Stéphane C,et al.Intraspecies diversity ofLactobacillussakeiresponse to oxidative stress and variability of strain performance in mixed strains challenges[J].Food Microbiology,2012,29(2):197-204.
[20] HIKMATE,NABIL B,ANTONIO C,et al.Characterization of lactic acid bacteria from naturally-fermented Manzanilla Alorea green table olives[J].Food Microbiology,2012,32(2): 308-316.
[22] 聂志强,韩玥,郑宇,等.宏基因组学技术分析传统食醋发酵过程微生物多样性[J].食品科学,2013, 34(15):198-203.
[23] YOSHIDA S,OKANO K,TANAKA T,et al.Homo-D-lactic acid production from mixed sugars using xylose-assimilating operon-integratedLactobacillusplantarum[J].Appl Microbiol Biotechnol,2011,92:67-76.
[24] VAN B de Veen H, ABEE T,TEMPELAARS M,et al.Short-and long-term adaptation to ethanol stress and its cross-protective consequences inLactobacillusplantarum[J].Appl Environ Microbiol,2011,77:5 247-5 256.
[25] LUCKWU de LUCENA BT,SILVA GG,MANOEL dos Santos B,et al.Genome sequences of the ethanol-tolerantLactobacillusvinistrains LMG 23202T and JP7.8.9[J].Journal of Bacteriology,2012,194(11):3 018-3 018.
[26] YE W,HUO G,CHEN J, et al.Heterologous expression of theBacillussubtilis(natto) alanine dehydrogenase inEscherichiacoliandLactococcuslactis[J].Microbiol Res,2010,165:268-275.
[27] SIEZEN RJ,BAYJANOV JR,FELIS GE,et al.Genome-scale diversity and niche adaptation analysis ofLactococcuslactisby comparative genome hybridization using multi-strain arrays[J].Microb Biotechnol,2011,4:383-402.
[28] PESSIONE A,LAMBERTI C,PESSIONE E.Proteomics as a tool for studying energy metabolism in lactic acid bacteria[J].Molecular Biosystems,2010,6(8):1 419-1 430.
[29] HAMON E,HORVATOVICH P,IZQUIERDO E,et al.Comparative proteomic analysis ofLactobacillusplantarumfor the identification of key proteins in bile tolerance[J].BMC Microbiol,2011,11(2):63.
[30] ANGEL TE,ARYAL UK,HENGEL SM,et al.Mass spectrometry-based proteomics:existing capabilities and future directions[J].Chem Soc Rev,2012,41:3 912-3 928.
[31] CARVALHO AL.Metabolic engineering ofLactococcuslactisfor improved tolerance to acid stress:guidelines frominvivoNMR analysis of glucose metabolism[D].Lisbon:New University of Lisbon,2012.
[32] LEVERING J,MUSTERS MW,BEKKER M,et al.Role of phosphate in the central metabolism of two lactic acid bacteria-a comparative systems biology approach[J].FEBS J,2012,279:1 274-1 290.
[33] MACHADO D,COSTA RS, FERREIRA EC,et al.Exploring the gap between dynamic and constraint-based models of metabolism[J].Metab Eng,2012,14(2):112-119.
[34] TENAZINHA N,VINGA S.A survey on methods for modeling and analyzing integrated biological networks[J]. IEEE/ACM Trans Comput Biol Bioinform,2011,8:943-958.
[35] SOH KC,MISKOVIC L,HATZIMANIKATIS V.From network models to network responses: integration of thermodynamic and knetic properties of yeast genome-scale metabolic networks[J].FEMS Yeast Res, 2012,12:129-143.
[36] REED JL.Shrinking the metabolic solution space using experimental datasets[J].Plos Computational Biology,2012,8(8):1 428-1 439.
[37] LERMAN J,HYDUKE DR,LATIF H,et al.In silico method for modelling metabolism and gene product expression at genome scale[J].Nat Commun,2012,3(1):929.
Advances in lactic acid bacteria genomics and its application
MA Chang-lu1, 2,PANG Xiao-yang1,ZHANG Shu-wen1, LIU Lu1,LU Jing1,LYU Jia-ping1*
1(Chinese Academy of Agricultural Sciences, Beijing 100193,China) 2(Beijing Agricultural Vocational College, Beijing 102442,China)
To better know the current advances in the lactic acid bacteria genomics and its application,a lot of literatures were collected and analyzed to find three key points in research, i.e.. Species identification, strain improvement (genetic recombination) and genomics. The results showed that the genomic studies of the mutants and specific strains of lactic acid bacteria, combined with proteomics and the related research results, would further reveal the essence of the functional characteristics of the lactic acid bacteria, and indicate that the lactic acid bacteria could be a member for cell factory, antiseptic function and health care.
lactic acid bacteria; species identification; genetic recombination; genomics
10.13995/j.cnki.11-1802/ts.201610040
博士研究生,副教授(吕加平为通讯作者, E-mail: kjdairy@126.com)。
国家自然科学基金面上项目(31471603);国家自然科学基金面上项目(31371808);北京农业职业学院科技研发推广类项目(XY-16-29)
2015-12-01,改回日期:2016-04-12
猜你喜欢
军事文摘(2021年18期)2021-12-02
现代畜牧科技(2021年9期)2021-10-13
成都医学院学报(2021年2期)2021-07-19
科学(2020年2期)2020-08-24
实用手外科杂志(2015年3期)2015-08-27
山西农经(2015年7期)2015-07-10
中国洗涤用品工业(2015年7期)2015-02-28
生物进化(2014年3期)2014-04-16
中国酿造(2014年9期)2014-03-11
食品工业科技(2014年15期)2014-03-11
