
Calcium channel inhibition-mediated axonal stabilization improves axonal regeneration after optic nerve crush
2016-12-01 09:23ViniciusT.Ribas,PaulLingor
中国神经再生研究(英文版) 2016年8期
Calcium channel inhibition-mediated axonal stabilization improves axonal regeneration after optic nerve crush
Axonal projections are specialized neuronal compartments and the longest parts of neurons. Axonal degeneration is a common pathological feature in many neurodegenerative disorders, such as Parkinson’s disease, amyotrophic lateral sclerosis, glaucoma, as well as in traumatic lesions of the central nervous system (CNS), such as spinal cord injury. In many neurological disorders, the axon is the first neuronal compartment affected, preceding the death of cell bodies. Following a lesion to the CNS, damaged axons degenerate and usually fail to regenerate past the point of the original injury, resulting in permanent deficits.
The process of axonal degeneration is a regulated self-destructing cellular mechanism, which involves different steps. In the spinal cord and optic nerve, a focal traumatic lesion to the axons results in a sudden axonal disintegration extending for about 500 μm on both sides of the lesion that is termed acute axonal degeneration (Knoferle et al., 2010). After the fast disintegration of the adjacent parts of the lesioned axon during acute axonal degeneration, the rest of the axon remains morphologically stable within the following hours. At later time points the distal part of the axon undergoes Wallerian degeneration characterized by a widespread breakdown of the axonal cytoskeleton, destruction of internal organelles and ultimately axonal disintegration, while the proximal part of the axon starts the so-called slow dying back. At the molecular level, the initial axonal injury leads to a rapid calcium influx into the axon. Downstream of calcium, calpain proteases, which are key mediators of cytoskeletal degradation, are activated. In addition to calpain activation, autophagy is another important mechanism downstream of calcium that is increased in the course of axonal degeneration in the optic nerve and the spinal cord (Knoferle et al., 2010; Ribas et al., 2015). Channel-mediated influx of extracellular calcium is critical for initiating acute axonal degeneration, as calcium channel blockers prevent the early intra-axonal rise in calcium and almost completely prevent the following axonal degeneration. Moreover, addition of a calcium ionophore significantly increases the speed of axonal disintegration (Knoferle et al., 2010). Therefore, calcium influx is an important priming process regulating axonal degeneration.
Numerous studies aiming at the improvement of outcome after traumatic axonal CNS lesions focused on neurorestorative approaches, such as stimulation of sprouting and axonal regeneration. The preservation of axonal integrity could be beneficial to improve such strategies. For example, increased axonal stabilization could lead to a shorter distance for the regenerating axons to regrow. Moreover, preserved and still connected axons, which would otherwise undergo secondary degeneration, could serve as guide structures for regenerating axons. Thus, failure to preserve axonal integrity could be one reason for limited functional recovery following traumatic lesions. However, it has not been systematically assessed whether the attenuation of axonal degeneration indeed improves the ability of axons to regenerate past a lesion site. Recently we addressed this question by blocking acute axonal degeneration using calcium channel inhibitors in a model of optic nerve crush (ONC) lesion and analyzing axon regeneration at later time points (Ribas et al., 2016).
The optic nerve injury model is a widely used paradigm, which offers the big advantage of an easy surgical access to the optic nerve itself and the vitreous permitting to target retinal ganglion cells (RGC) in order to assess their survival and regenerative properties. Our group showed previously, by optic nerve live-imaging experiments, that topical application on the optic nerve of a combination of the two calcium channel inhibitors (L-/N-type channel blocker amlodipine, T-type channel blocker amiloride) and the AMPA receptor blocker NBQX was able to block calcium influx and almost completely stabilize superficial axons after crush lesion (Knöferle et al., 2010).
We attempted to stabilize the maximum number of optic nerve axons by using a dual strategy to deliver calcium channel inhibitors to RGC axons: intravitreal injection and topical application on the optic nerve (Ribas et al., 2016). We found that our strategy was able to almost completely prevent the acute axonal degeneration of superficial axons after ONC assessed by in vivo live-imaging, corroborating previous results of our group. We additionally showed axonal stabilization localized in deeper regions of the optic nerve, although complete axonal protection in the inner optic nerve was not achieved. This incomplete axonal protection in deeper regions can be explained because superficial axons are more easily reachable by topical inhibitor application than the axons in the inner optic nerve. In addition, traumatic lesions can induce an increase in intraaxonal calcium concentration via different mechanisms, including influx from extracellular sources through mechanopores, as well as from intracellular stores such as mitochondria or the endoplasmic reticulum. Thus, this strategy might not completely block the rise in intraaxonal calcium concentration in all lesioned optic nerve axons.
It has been previously established that preventing calcium influx after traumatic lesion protects axons from degeneration. However, experiments that addressed the question of whether the specific blockage of acute axonal degeneration by calcium channel inhibition facilitates subsequent axonal regeneration distal to the lesion site were missing. We now showed that axonal stabilization by calcium channel inhibition significantly increases axon regeneration up to 2-fold distal to the crush lesion site, thus confirming this hypothesis. However, the increase in axonal regeneration was limited to the area close to the crush site, at larger distances from the crush site (≥ 400 μm), the treatment was not effective. In the adult CNS, a lesion to the axons results in axonal degeneration and the axons fail to regenerate past the point of the original injury. The failure in the regenerative response of adult CNS neurons is predominantly caused by the weak intrinsic growth capacity of adult neurons and the presence of growth-repressing molecules in the CNS environment. In our study we did not target the inhibitory environment neither the intrinsic capabilities for axonal outgrowth. Thus, the effect we observed on axonal regeneration is only due to the increased axonal stabilization. In conclusion, our proof-of-principle study showed that axonal stabilization by inhibition of calcium channels facilitates axonal regeneration and it could be combined with additional strategies in order to elicit a more robust effect.
In addition to axonal protection, promoting cell survival is essential for successful regeneration. We therefore also evaluated neuronal survival and found that inhibition of calcium channels increases RGC survival after ONC. We targeted AMPA receptors and these receptors have been linked to excitotoxic neuronal death which involves increased calcium influx. In addition, previous studies also found that inhibition of calcium channels increased RGC survival. Now, our study showed that in addition to the effect on RGC survival, calcium channel inhibition decreases axonal degeneration and improves axonal regeneration. These effects mediated by calcium channel inhibition seem to be specific and were not observed in previous studies targeting other mechanisms involved in axonal degeneration. For example, the Wallerian degeneration (Wlds) mutation, which protects axons from degeneration through a completely different mechanism, does not increase RGC survival (Beirowski et al., 2008). Thus, our study points to calcium channels as therapeutic targets, which in addition to axonal protection also regulate RGC survival.
We next evaluated the molecular downstream cascade involved in the effects of calcium channel inhibition. Here we found that inhibition of calcium channels reduces calpain activity in the lesioned optic nerve. Calpain is a calcium-dependent protease, which cleaves a variety of vital cellular components. Calpain inhibitors protect axons from degeneration, indicating that calpain activity is important for axonal degeneration. Moreover, calpain inhibition has a neuroprotective effect against axonal damage-induced RGC death. Thus, the reduction of calpain activity by calcium channel inhibition could provide a mechanistic link to the effect on axonal degeneration and RGC survival. We also found that the activity of the c-Jun N-terminal kinase (JNK)/c-Jun signaling pathway was attenuated by inhibition of calcium channels. JNK is a serine/threonine kinase that regulates RGC death and axon degeneration after optic nerve lesion. The tran-scription factor c-Jun is the major target of JNK and regulates RGC apoptosis after optic nerve lesion. The activation of JNK/c-Jun signaling pathway can be activated by a variety of cellular stresses. For example, ONC induces activation of JNK signaling pathway in RGC via TNFα. Our study now points to calcium influx as an additional mechanism involved in activation of the JNK/c-Jun signaling pathway. Therefore, decreased activity of JNK/c-Jun by calcium channel inhibition could be an additional mechanism contributing to attenuated axonal degeneration and RGC death. Finally, we showed that the activation (phosphorylation) of the pro-survival serine/threonine protein kinase Akt was increased in the ganglion cell layer of retinas treated with calcium channel inhibitors. Akt has a pivotal function in mediating survival signaling in neuronal cells, as well as, axonal outgrowth, including in RGC. Thus, the increase in Akt activation induced by calcium channel inhibition could explain the effects on RGC survival and axonal regeneration (Figure 1).
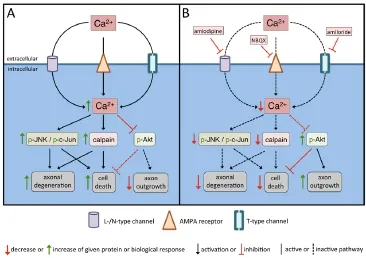
Figure 1 Scheme of the proposed cellular and molecular effects of calcium channel inhibition on RGC cell body and axons after optic nerve crush (ONC).
Several previous studies focused on axonal stabilization after traumatic injury to the CNS. For example, the Wlds mutation or the expression of nicotinamide mononucleotide adenylyltransferase 3 both decrease axonal degeneration, but axon regeneration was not evaluated here (Beirowski et al., 2008; Kitaoka et al., 2013). Taxol, a microtubule stabilization agent, significantly stabilizes axons after traumatic injury to the CNS acting directly on the affected cytoskeleton (Ertürk et al., 2007). More recently, Hellal et al. described robust increase in axon regeneration by Taxol treatment in a model of dorsal spinal cord injury (Hellal et al., 2011). In addition, Taxol increases axon regeneration after optic nerve crush lesion, but did not influence the survival of RGCs (Sengottuvel et al., 2011). Although, axonal stabilization partially accounts for the effect of Taxol on axon regeneration, decreased fibrotic and glial scar formation also contribute to it (Hellal et al., 2011; Sengottuvel et al., 2011). Moreover, a recent study trying to reproduce the data from Hellal et al. (2011) could not observe an increase in axonal regeneration despite showing a decrease in fibrotic scar formation (Popovich et al., 2014). Our study now showed that calcium channel inhibition-mediated axonal stabilization improves RGC survival and axonal regeneration. Although calcium influx is a very rapid event after traumatic lesion to the CNS, even delayed blocking of calcium influx was shown to decrease secondary axon loss after a contusive spinal cord injury (Williams et al. 2014). Thus, our study contributes to an improved understanding of the value of calcium influx blockers in the limitation of axonal degeneration and neuronal death, as well as improved axonal regeneration. In a translational approach, additional studies will be required to titrate the optimal dosage, the exact timing, the best localization to apply the treatment and any to implement combinatorial strategies.
Taken together, in our study, using a rat ONC model, we found that application of calcium channel inhibitors preserved axonal integrity from acute degeneration and consecutively increased survival of RGCs and improved axonal regeneration. Moreover, we showed that calcium channel inhibitors decreased lesion-induced calpain activation, attenuated the activation of the JNK/c-Jun signaling pathway and increased the activation of the pro-survival kinase Akt, suggesting that these mechanisms could be involved in the effects of calcium channel inhibitors. In conclusion, our study shows that an intervention targeting axonal integrity could be an important step in a combinatorial therapeutic strategy to promote functional recovery after traumatic injury to the CNS and points to calcium channel inhibitors as valuable therapeutic agents in CNS trauma.
This study is funded by a fellow of the Coordination for the Improvement of Higher Education Personnel (CAPES), Brazil to VTR, and a funding from the DFG-Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB) to PL.
Vinicius T. Ribas, Paul Lingor*
Brain Institute, Federal University of Rio Grande do Norte, Natal,
Brazil (Ribas VT)
Department of Neurology, University Medicine Göttingen, Göttingen,
Germany (Ribas VT, Lingor P)
Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), Göttingen, Germany (Lingor P)
*Correspondence to: Paul Lingor, M.D., plingor@gwdg.de.
Accepted: 2016-08-03
orcid: 0000-0001-9362-7096 (Paul Lingor)
How to cite this article: Ribas VT, Lingor P (2016) Calcium channel inhibition-mediated axonal stabilization improves axonal regeneration after optic nerve crush. Neural Regen Res 11(8):1245-1246.
References
Beirowski B, Babetto E, Coleman MP, Martin KR (2008) The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci 28:1166-1179.
Ertürk A, Hellal F, Enes J, Bradke F (2007) Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci 27:9169-9180.
Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331:928-931.
Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, Takagi H (2013) Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis 4:e860.
Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Bruck W, Bahr M, Lingor P (2010) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 107: 6064-6069.
Popovich PG, Tovar CA, Lemeshow S, Yin Q, Jakeman LB (2014) Independent evaluation of the anatomical and behavioral effects of Taxol in rat models of spinal cord injury. Exp Neurol 261:97-108.
Ribas VT, Koch JC, Michel U, Bähr M, Lingor P (2016) Attenuation of axonal degeneration by calcium channel inhibitors improves retinal ganglion cell survival and regeneration after optic nerve crush. Mol Neurobiol doi: 10.1007/s12035-015-9676-2.
Ribas VT, Schnepf B, Challagundla M, Koch JC, Bähr M, Lingor P (2015) Early and sustained activation of autophagy in degenerating axons after spinal cord injury. Brain Pathol 25:157-170.
Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D (2011) Taxol facilitates axon regeneration in the mature CNS. J Neurosci 31:2688-2699.
Williams PR, Marincu BN, Sorbara CD, Mahler CF, Schumacher AM, Griesbeck O, Kerschensteiner M, Misgeld T (2014) A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat Commun 5:5683.
10.4103/1673-5374.189184

- 中国神经再生研究(英文版)的其它文章
- Secondary parkinsonism induced by hydrocephalus after subarachnoid and intraventricular hemorrhage
- Prospects for bone marrow cell therapy in amyotrophic lateral sclerosis: how far are we from a clinical treatment?
- Uncoupling protein 2 in the glial response to stress: implications for neuroprotection
- Selective neuronal PTEN deletion: can we take the brakes off of growth without losing control?
- TRPV1 may increase the effectiveness of estrogen therapy on neuroprotection and neuroregeneration
- Tamoxifen: an FDA approved drug with neuroprotective effects for spinal cord injury recovery