
固相萃取–气相色谱–串联质谱法测定茶叶中9种农药残留量*
2016-11-30 08:14颜鸿飞王美玲陈练吕小园戴洁芸
化学分析计量 2016年6期
颜鸿飞,王美玲,陈练,吕小园,戴洁芸
(湖南出入境检验检疫局,国家食品安全检测重点实验室,长沙 410004)
固相萃取–气相色谱–串联质谱法测定茶叶中9种农药残留量*
颜鸿飞,王美玲,陈练,吕小园,戴洁芸
(湖南出入境检验检疫局,国家食品安全检测重点实验室,长沙 410004)
建立固相萃取净化–气相色谱–串联质谱法同时测定茶叶中9种农药残留量的方法。茶叶样品用乙腈均质提取,提取液经固相萃取净化处理后,采用DB–5MS毛细管色谱柱分离,在多反应监测模式下测定,外标法定量。9种农药组分的质量浓度在0.01~0.50 mg/L范围内与其色谱峰面积呈良好线性,相关系数r2大于0.998,方法测定下限(10 S/N)为0.002~0.01 mg/kg。以空白绿茶、红茶、普洱茶和乌龙茶为基体,在0.05,0.1,0.2 mg/kg 3个添加水平进行加标回收试验,加标回收率在73.6%~99.7%之间,相对标准偏差为4.2%~8.7%(n=6)。该法操作简便、快速,适用于茶叶中多种农药残留的测定。
固相萃取;气相色谱–串联质谱法;茶叶;农药残留
茶叶是我国主要的饮品之一,我国是茶叶生产、消费和出口大国。近年来,茶叶中农药残留问题受到了人们的广泛关注,主要茶叶进口国家和地区纷纷制定和实施越来越严格的茶叶农药残留新标准,给我国茶叶出口带来了巨大的冲击和考验。香港《食物内除害剂残余规例》于2014年8月1日起正式实施,该技术标准对茶叶中10余种除害剂进行了重点监控[1-2]。与此同时,冗长且繁琐的农残检测样品前处理技术已难以适应新形势下茶叶农残日常检测工作需求。因此建立茶叶中多种农药残留的快速、简便、准确的检测方法具有重要的实际意义。常见茶叶中农药残留的分析方法有气相色谱法(GC)[3–6]、气相色谱–质谱法(GC–MS)[7–9]、气相色谱–串联质谱法(GC–MS–MS)[10]、液相色谱–串联质谱法(LC–MS–MS)[11–16]等。其中GC–MS–MS分离性能高,可同时对上百种农药进行快速定性、定量检测,比单级四级杆有更高的灵敏度和抗干扰能力,在农残检测领域中的应用越来越广泛。固相萃取法(SPE)作为新型样品处理技术,具有简单、快速、试剂消耗小、成本低等优点,已广泛应用于食品中农药残留分析[5–8]。笔者针对香港《食物内除害剂残余规例》中茶叶重点监控的农残检测项目,用乙腈均质提取茶叶样品,经石墨化碳固相萃取小柱净化,采用GC–MS–MS法测定,实现了茶叶中9种农药残留量的快速测定。
1 实验部分
1.1 主要仪器与试剂
气相色谱质谱/质谱联用仪:Varian 300型,美国瓦里安公司;
真空旋转蒸发仪:LABOROTA 4003型,德国海道夫公司;
全自动均质仪:Autogizer 1型,美国Tomtec公司;
乙腈、丙酮、正己烷:色谱纯,美国Tedia公司;
无水硫酸钠:分析纯,上海国药集团化学试剂有限公司;
乙酰甲胺磷、莠灭净、二嗪磷、α-六六六、β-六六六、γ-六六六、δ-六六六、杀扑磷、丙溴磷标准品:纯度均大于90%,德国Dr.Ehrenstorfer公司;
石墨化碳黑固相萃取柱:0.5 g,6 mL,美国安捷伦公司;
实验用水为超纯水。
1.2 仪器工作条件
1.2.1 气相色谱条件
色谱柱:DB–5MS石英毛细管柱(30 m×0.25 mm,0.25μm,美国安捷伦科技有限公司);柱温:100℃保持1 min,以20℃/min升温至300℃,保持5 min;载气:氦气,纯度不小于99.999 %,流量为1.0 mL/min;进样口温度:250℃;进样体积:1 μL;不分流进样;GC–MS–MS接口温度:280℃。
1.2.2 质谱条件
扫描方式:多反应监测模式(MRM),每种化合物分别选择一个母离子,2个子离子,每组所有需要检测的离子按照出峰顺序,分时段分别检测;电离方式:电子轰击离子源(EI),70 eV;离子源温度:250℃;循环时间:0.2 s;色谱过滤峰宽:3.0 s;碰撞气:氩气,纯度不低于99.999%;碰撞气压力:0.2 Pa。
9种化合物的保留时间、定性离子对、定量离子对、碰撞能量见表1。
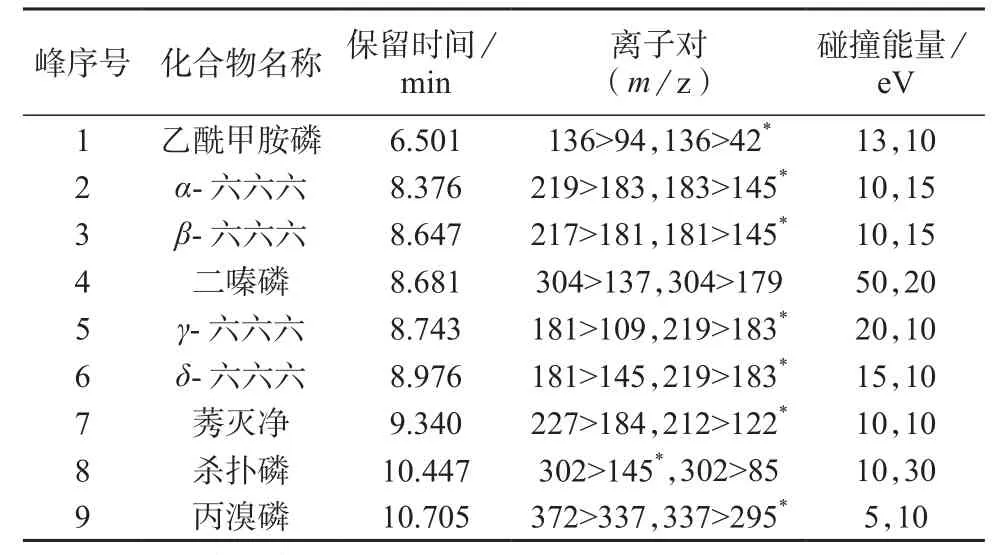
表1 9种农药的保留时间、定量离子、定性离子、碰撞能量
1.3 样品预处理
取有代表性的茶叶样品500 g,经粉碎机粉碎,过0.83 mm(20目)筛,混匀,均分成两份作为试样,分装入洁净的容器内,密封并标记。制存样过程中,应防止样品受到污染或发生残留物含量的变化。试样于常温下保存。
称取1.0 g (精确至0.01 g)试样于50 mL具塞聚四氟乙烯离心试管中,加入10 mL乙腈,以15 000 r/min转速均质提取2 min,再以6 000 r/min离心3 min,将上层清液转移至100 mL鸡心瓶中。残渣用10 mL乙腈重复均质提取一次,离心后,取上层清液,合并两次提取液于同一100 mL鸡心瓶中,于40℃水浴中旋转蒸发至近干,加入2 mL丙酮–正己烷溶液(体积比为1∶1,下同)溶解残渣,待净化。将石墨化碳黑固相萃取柱置于固相萃取装置上,在柱中加入2 cm高的无水硫酸钠,依次用3 mL丙酮、6 mL丙酮–正己烷溶液(1∶1)预淋洗,弃去淋洗液,当液面到达无水硫酸钠层顶部时,将以上得到的试样提取液转入石墨化碳黑固相萃取柱中,然后用12 mL丙酮–正己烷溶液(1∶1)洗涤鸡心瓶,并将洗涤液全部加到石墨化碳黑固相萃取柱中,收集全部流出液于20 mL刻度离心管中,在40℃下用平缓氮气流吹至近干,用丙酮溶解并定容至1.0 mL,供GC–MS–MS 测定。同样做基质空白溶液并测定。
1.4 标准溶液配制
标准储备液:分别准确称取0.1 g(精确至0.1 mg)乙酰甲胺磷、莠灭净、二嗪磷、α-六六六、β-六六六、γ-六六六、δ-六六六、杀扑磷和丙溴磷农药标准品于100 mL棕色容量瓶中,用丙酮溶解并定容至标线,摇匀,配制成质量浓度为1 mg/mL标准储备液,在0~4℃避光保存。
混合农药标准储备液:准确吸取乙酰甲胺磷、莠灭净、二嗪磷、α-六六六、β-六六六、γ-六六六、δ-六六六、杀扑磷和丙溴磷标准储备溶液各1.0 mL于100 mL棕色容量瓶中,用丙酮溶解并定容至标线,配制成质量浓度均为10 µg/mL的混合农药标准储备液,于0~4℃避光保存。
基质标准工作溶液:准确吸取一定体积的混合农药标准储备液,用基质空白溶液逐级稀释成质量浓度分别为10,20,50,100,200,500 µg/L的系列基质标准工作溶液,现配现用。
2 结果与讨论
2.1 萃取溶剂的选择
食品中农残分析需要在复杂基质中对目标化合物进行选择性提取、富集和分离,特别是在多组分残留分析中,目标农药品种不同,物理化学性质存在差异,选择合适的提取溶剂往往在分析中起着重要作用。在选择萃取溶剂时,不仅要考虑目标分析物在溶剂中的溶解度、溶剂与基质的浸透效果,还需考虑不同萃取溶剂对萃取效率的影响。目前,提取溶剂主要有丙酮、甲醇、乙酸乙酯、乙腈和正己烷等及其混合溶剂[10]。实验表明,用丙酮和甲醇对大多数农残的提取效果好,但同时提取的杂质多,给后续净化操作带来难度;二氯甲烷和正己烷对茶叶组织渗透力不够,难以将极性农药从植物组织中完全萃取出来,提取效率较低;用乙腈提取,组织渗透力强,极性大小较合适,对大多数农药有较高提取效率,同时色素和其它弱极性杂质提取较少,已在食品多农残分析中被广泛采用。因此本实验采用乙腈作为提取溶剂。
2.2 萃取方式的选择
常用的样品提取方法有液液萃取法[11,13]、均质提取法[4–5,14,16]、超声提取法[15]和固相萃取法[8–9,12]等。试验对比振荡提取、超声提取、均质提取等3种提取方法的提取效率,结果见图1。由图1可知,茶叶样品采用振荡和超声提取对目标农药中极性较大的农药的提取效率高,但是对于极性弱的六六六等农药提取效率较低,而采用高速均质提取对绝大多数农药都可完全提取,故本实验选择均质提取法。
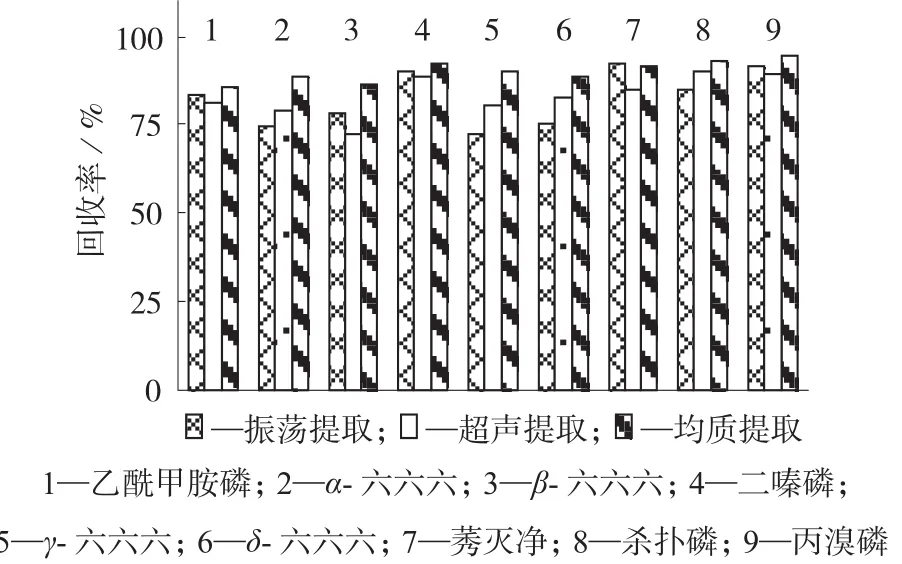
图1 不同提取方式下农药的提取回收率
2.3 固相萃取净化条件的选择
茶叶中含有大量的色素、生物碱、多酚类、氨基酸等杂质,给样品净化操作带来一定的难度。农残分析样品净化方法主要有液–液分配法[3,7]、渗透凝胶色谱[9]和固相萃取法[4–8]等。液–液分配净化是最简单的传统净化方式,基于被测农药具有相似溶解特性的前提条件,不适用于化学性质差异较大的多种农药残留的提取;常规凝胶渗透色谱(GPC)净化更适合富含油脂的样品,但存在仪器昂贵、耗时长和试剂成本高等问题;固相萃取柱净化是现代农药残留分析中新型的净化技术,在食品农残分析检测领域已得到广泛应用[4–5,7–8]。对几种农残分析中常用的固相萃取柱如石墨化碳、中性氧化铝、弗罗里硅土、石墨化碳/氨基等的净化效果进行比较,实验发现,石墨化碳、石墨化碳/氨基固相萃取柱的净化效果都较好,但考虑到石墨化碳/氨基固相萃取柱成本相对较高,且洗脱试剂较复杂。故本实验采用石墨化碳固相萃取柱。
2.4 气相色谱–质谱条件的选择
通过反复调整进样口温度、色谱柱温度、载气流速等气相色谱关键参数,保证所有的样品基质中目标农药有较好的分离度,尽量避免基质干扰峰与目标化合物保留时间的重叠,为提高分析效率,尽量缩短分析时间,最后得到合适的色谱柱程序升温条件:于100 ℃保持1 min,然后以20℃/min程序升温至300℃,保持5 min;色谱柱选用DB–5MS石英毛细管柱(30 m×0.25 mm,0.25μm)进行分离;载气流速为1.0 mL/min。在优化的气相色谱条件下,采用Q3 SCAN全扫描方式得到各待测农药的质谱图,然后选取2~3个相对丰度较高、质荷比较大及相同扫描时间段其它农药及无背景干扰的离子作为母离子、子离子对,再将各农药成分的扫描分时间段,进一步优化碰撞能量,以保证检测的灵敏度和准确性。9种农药的保留时间和MRM离子参数见表1,标准物质溶液的MRM总离子流图见图2。 2.5 方法的线性和检出限
图2 9种农药标准溶液的MRM总离子流图
茶叶成分较复杂,基质效应较明显,为补偿样品基质对定量分析的影响,采用基质匹配校准混合溶液及建立基质匹配校准曲线进行定量。取1.4制备的系列基质标准工作溶液,按照1.2仪器工作条件进行测定,以各自定量离子对的峰面积(y)对质量浓度(x,µg/L)作基质匹配校准曲线和线性回归分析,得到各农药的线性方程和相关系数,见表2。由表2可知,在10~500 µg/L质量浓度范围内,9种农药的基质匹配校准曲线的线性相关系数r2均大于0.998,线性关系良好。用茶叶空白基质样液逐级稀释混合标准溶液,以信噪比S/N=10确定各组分的测定下限,结果见表2。由表2可知,本方法的灵敏度满足香港地区规例对茶叶中目标农药的最新残留限量标准要求。

表2 9种农药的线性回归方程和相关系数及测定下限
2.6 方法的回收率和精密度
采用标准加入法,在不含检测目标物的空白茶叶样品中分别添加0.05,0.1,0.2 mg/kg 3个浓度水平的标准混合溶液进行加标回收试验,在添加溶液与样品充分浸润混匀,溶剂挥发完全后进行测试,每个水平重复6次,计算添加回收率和相对标准偏差。红茶空白样品、红茶加标样品的MRM总离子流色谱图见图3,实验结果见表3。
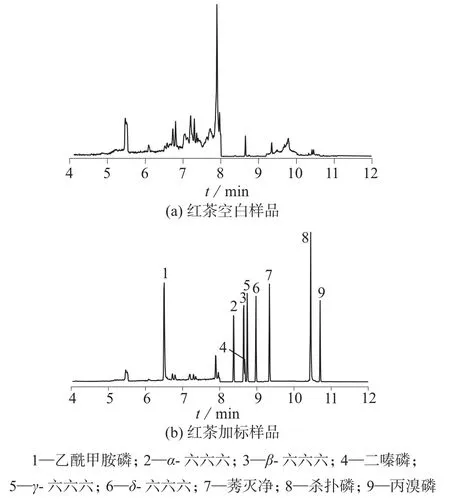
图3 样品的MRM总离子流图
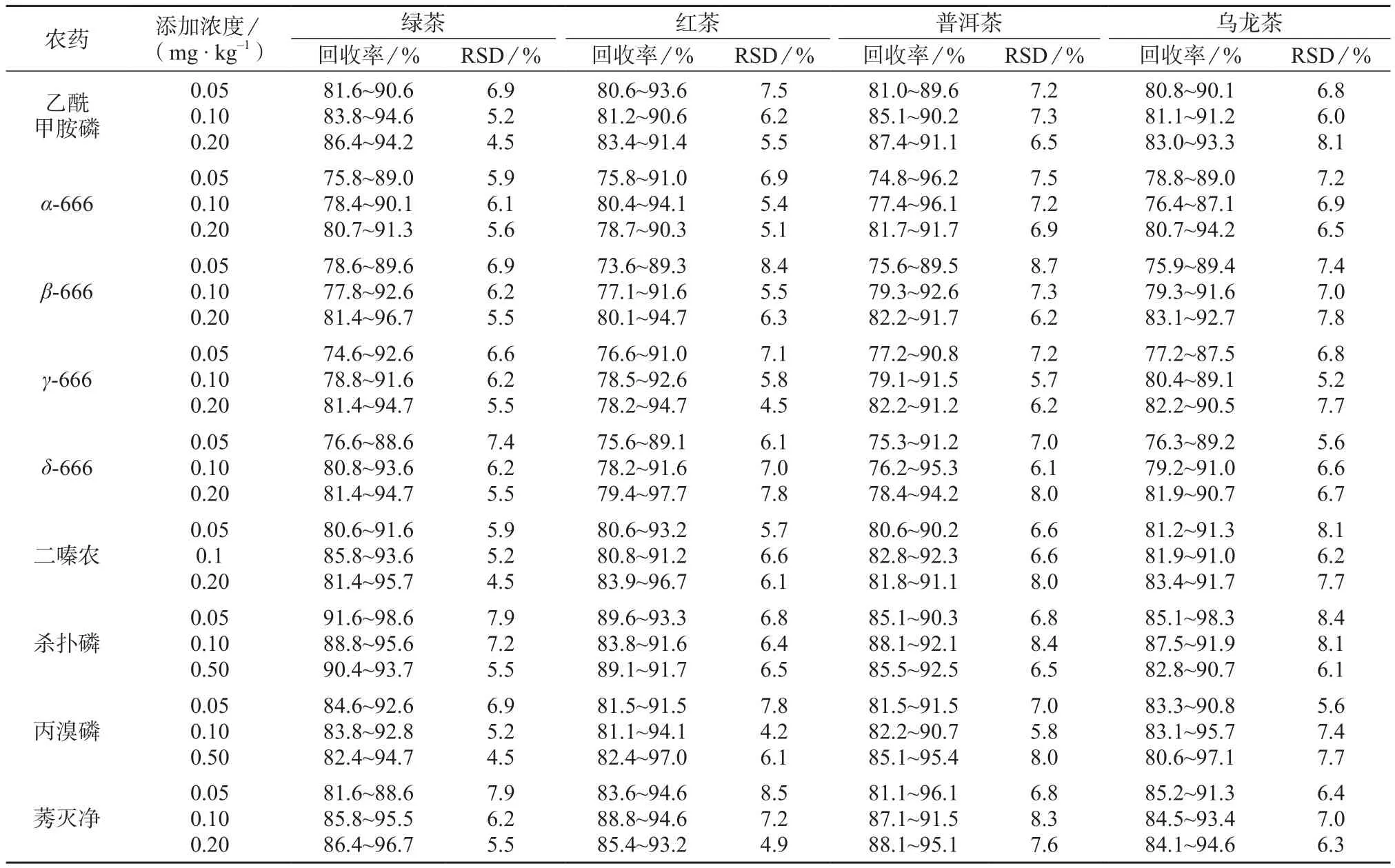
表3 茶叶样品加标回收率和精密度试验结果(n=6)
由表3可知,9种农药的加标回收率为73.6%~99.7%,测定结果的相对标准偏差为4.2%~8.7% (n=6),各技术指标满足多农残检测标准要求。
3 结语
通过对茶叶样品进行均质萃取、固相萃取净化等样品前处理,建立了茶叶中9种农药残留量的气相色谱–串联质谱测定方法。该方法操作简便、快速、准确,灵敏度高,可满足出口茶叶中多种农药残留日常检测工作需要。
[1] 童小麟,陈建民.我国出口欧盟茶叶农药残留超标通报的分析与应对[J].检验检疫学刊,2015,25(1): 16–18.
[2] 孙正君,汤桦,陈大舟,等.茶叶中农药最大残留限量标准及检测方法比较[J].计量学报,2010,31(z1): 121–124.
[3] 廖和菁,张雪春,胡礼渊,等.气相色谱法同时测定茶叶中10 种有机磷农药残留[J].中国食品卫生杂志,2015,27(1): 41–44.
[4] 郗艳丽,刘敏,李雅玲,等.固相萃取气相色谱法测定茶叶中5种有机磷农药残留[J].吉林医药学院学报,2016,37(1): 7–10.
[5] 吴林,吴晓波,张承聪,等.固相萃取–气相色谱法测定茶叶中多氯联苯和有机氯农药残留[J].云南化工,2011,38(2): 21–24.
[6] 靳保辉,陈沛金,谢丽琪,等.茶叶中25种有机氯农药多残留气相色谱测定方法[J].分析测试学报,2007,26(1): 104–106.
[7] GB/T 23204–2008 茶叶中519种农药及相关化学品残留量的测定 气相色谱–质谱法[S].
[8] 罗逢健,陈宗懋,汤富彬,等.固相萃取和气相色谱质谱法测定茶叶中34 种农药残留[J].农药,2010,49(5): 363–366.
[9] 卢大胜,邱欣磊,林元杰,等. QuEnChERS 方法联合GPC–GC–MS测定蔬菜水果中农药残留中的应用[J].质谱学报,2011,32(4): 229–235.
[10] 沈伟健,余可垚,桂茜雯,等.分散固相萃取–气相色谱–串联质谱法测定蔬菜中107种农药的残留量[J].色谱,2009,27(4): 391–400.
[11] 廖国会,金星,陈文龙.高效液相色谱法同时检测茶叶中吡虫啉和啶虫脒残留的方法[J].贵州农业科学,2011,39(4): 130–132.
[12] 陈红平,刘新,王川丕,等.超高压液相色谱–串联质谱测定茶叶中10种极性农药残留[J].分析试验室,2011,30(8): 48–53.
[13] 郭防.茶叶中农药残留检测的样品前处理研究进展[J].微量元素与健康研究,2012,29(1): 61–64.
[14] 杨方,杨守深,林永辉,等.超高效液相色谱–电喷雾电离串联质谱联用法检测茶叶中阿维菌素类药物残留[J].色谱,2009,27(2): 153–157.
[15] 谢文,钱艳,丁慧瑛,等.液相色谱–电喷雾电离三级四极杆质谱法测定茶叶中6种烟碱类农药残留[J].分析化学,2009,37(4): 495–499.
[16] 陈琼,喻芳,张惠,等. QuEChERS法处理结合液质法测茶叶中多种农残[J].广州化工,2015,43(16): 143–146.
Determination of Nine Pesticides Residues in Tea by Solid Phase Extraction and Gas Chromatography–Tandem Mass Spectrometry
Yan Hongfei, Wang Meiling, Chen Lian, Lyu Xiaoyuan, Dai Jieyun
(Hunan Entry–Exit Inspection and Quarantine Bureau, State Key Laboratory of Food Safety Testing, Changsha 410004, China)
A gas chromatography–tandem mass spectrometry(GC–MS–MS) method was developed for simultaneous determination of nine pesticides residues in tea. The samples were extracted with acetonitrile and cleaned up using solid phase extraction. After separated on DB–5MS capillary column,the target analytes were detected by GC–MS–MS under the multi reaction monitoring mode and quantified by the external standard method. The correlation coefficents (r2) of nine pesticides were more than 0.998 obtained within the concentration range of 0.01 to 0.5 mg/L. The limits of quantitation varied from 0.002 to 0.01 mg/kg. At three spiked levels of 0.05, 0.1, 0.2 mg/kg in blank tea, the average recoveries ranged from 73.6% to 99.7% and the relative standard deviations were from 4.2% to 8.7% (n=6). The method is easy to operate and fast, and it is suitbale for determination of pesticides residues in tea.
solid phase extraction; gas chromatography–tandem mass spectrometry; tea; pesticides residues
O657.7
A
1008–6145(2016)06–0062–05
10.3969/j.issn.1008–6145.2016.06.015
*湖南省科技计划项目(2015JC3128)
联系人:颜鸿飞;E-mail: yanhf@hnciq.gov.cn
2016–10–17
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
理化检验-化学分册(2020年12期)2021-01-26
当代水产(2019年3期)2019-05-14
食品界(2018年8期)2018-09-03
西南石油大学学报(自然科学版)(2018年1期)2018-02-10
Coco薇(2017年7期)2017-07-21
金色年华(2016年23期)2016-06-15
铁道科学与工程学报(2015年5期)2015-12-24
质谱学报(2015年5期)2015-03-01
油气地质与采收率(2014年6期)2014-12-16
