
DNA片段分选微流控芯片发展现状
2016-11-19 19:17李哲煜孙凯张笑颜刘绍琴江雷任南琪
分析化学 2016年4期
李哲煜 孙凯 张笑颜 刘绍琴 江雷 任南琪
摘 要 新一代测序速度越来越快,费用不断降低,但整个流程中还有不少瓶颈。最耗时且影响测序准确度和重复性的环节之一,就是在文库制备过程中,对打断的大量基因组DNA片段进行基于传统凝胶电泳的手工操作筛选。近几年市场上出现了几种目标片段自动分选仪器,并迅速被多家国际知名测序中心引入测试,从而引起了广泛关注。本文评述了DNA片段分选发展经历,包括凝胶电泳、毛细管电泳、特别是微流控芯片在分选精度和通量上的不断提升,并简要论述了DNA分选目前尚存在的问题。
关键词 毛细管电泳; 微流控芯片; 片段分选; DNA测序; 基因表达; 综述
1 引 言
近年来,通过污染物对生物细胞水平的毒理学机理研究,以及对基因表达、蛋白表达和代谢产物产生影响的机理研究,并建立它们之间的关系,实现疾病的早期预防、诊断和治疗成为研究热点[1,2]。因此,基因表达和测序实验量不断增大,从而对提升实验仪器自动化效率的需求也随之增加。围绕这一背景需求,各国政府均投入巨额资金以应对新一轮的环境危机和对生命体健康以及未来人类生存的影响,由此产生的高科技核心技术以及相关产品市场化竞争日趋激烈。其中,测序技术和基因表达分析技术领域得到了空前的发展。
新一代测序技术虽然大大提高了测序速度,降低了测序费用,但整个流程中仍然存在不少瓶颈,限制了它的通量。其中,在文库制备过程中对打断的基因组DNA片段进行手工筛选(分选,即分离+目标筛选)就是重要瓶颈之一。
本文论述了DNA片段分选从凝胶电泳、毛细管电泳,到微流控芯片电泳的发展历程与各自的技术特点,并特别阐述了DNA片段的仪器化发展以及未来发展前景。
2 凝胶电泳与切胶回收
目前,DNA片段分选普遍使用的是基于凝胶电泳分离的筛选方法(手工切胶分选法),即从凝胶电泳分离后的琼脂糖凝胶或者聚丙烯酰胺凝胶中切胶回收,然后加热溶解、纯化目标片段。这种方法原理简单、材料易于获得、操作相对简单、成本低、回收量大,所以至今仍被广泛使用。但是,该方法凝胶制备准备时间长、消耗人力、不易扩展;最重要的是,源自焦耳热引起的扩散效应导致识别/分离精度低、分选准确性差。分选的准确性主要指,不同运行之间、不同操作员之间的结果存在差异,基于手工操作的凝胶分选通常会有10%以上来自样品之间交叉污染的纯度误差。操作至少需要15 s/片段。因此,对于大量重叠的片段分选,实验人员的手工操作很难保证准确度和效率。
目前,在传统凝胶电泳分选基础上发展出一些新方法,如冻融离心法[1]、凝胶浸泡法[2]、改良硅藻土法[3]、电泳洗脱法、石英棉纯化法[4]和海绵吸收法[5]。但是,这些方法并没有解决凝胶电泳固有的分离精度和分选准确度问题,操作仍然繁琐、难以实现自动化控制、回收耗时长,如果采用专用的回收试剂盒,所需费用较贵。因此迫切需要高精度、高效率分选法以及相应的高度自动化分选仪器来解决这个关键的瓶颈问题。
3 基于毛细管电泳与色谱技术的自动分选
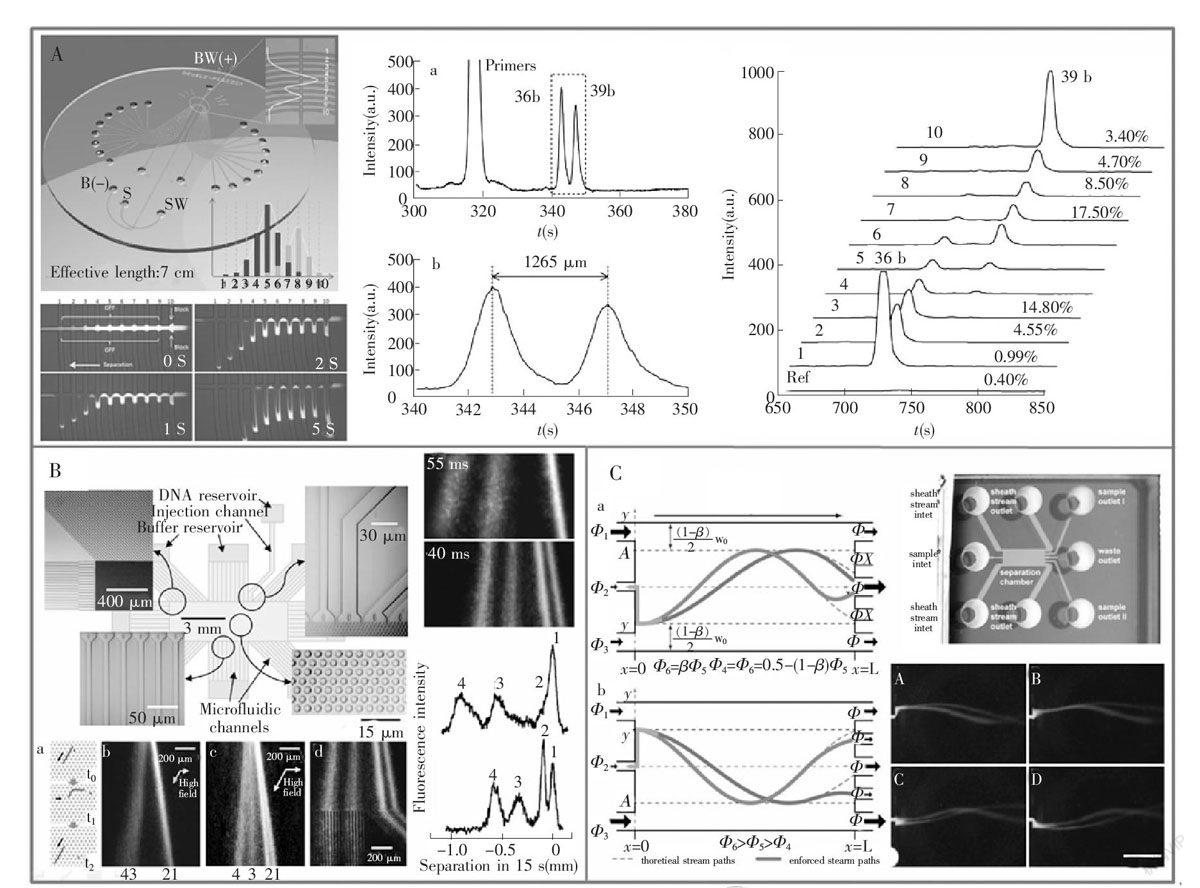
毛细管电泳技术是20世纪80年代后期迅速发展起来的一种分离分析技术[5,6]。利用毛细管电泳技术替代凝胶电泳节省了时间,提高了分离精度。但是,目标样品的分选仍然是个问题。Biofocus公司在毛细管电泳后加装了馏分收集装置来解决这个问题(图1a)[7],所得到的最好分选精度达到3个碱基差。但是,每次的操作仍然很复杂,必须先做一次或多次的分析操作,以确定每一种物质的迁移时间,并调整分离条件,使目的组分的峰之间能很好地分离,保证至少10~20 s的间隔;另外,采用的机械收集单元增大了死体积。由于毛细管电泳的检测窗口和样品的回收位置也就是毛细管的末端相距有一段距离(通常10 cm左右),所以目标片段到达收集出口的时间很难准确确定。Minarik等[8]为此在毛细管末端设计了两个UV检测器,以准确计算流经片段的速度(图1b)。但是如果检测点与回收出口不在同一点,误差将是不可避免的。
为此,日立公司的研究人员在阳极回收端设计了一个石英池,在石英池内为分离用的毛细管与收集用的毛细管之间保留了1 mm距离,检测点就设置在这个1 mm的间隙处(图1c)[9]。当DNA片段泳动出分离毛细管,分离电场被检测信号触发而断开,DNA片段随即利用鞘流(Sheathflow)进入12 cm长的收集毛细管,最终回收至样品管中。虽然该装置的回收精度得到很大提高,但是为了达到1个碱基差的分选精度,仍需要2次PCR扩增和共3次的分离回收。一个原因归结为1 mm的间隙仍然太大;另一个原因是,为了收集目标样品而中断分离电场导致了样品断片扩散。Huge等[10]也引入压力驱动流作为鞘流,更有效地抑制了回收端的交叉污染;并加快了回收端目标物的迁移速度,最后通过毛细管末端出口的喷嘴以液滴的形式点样到96孔板,从而提高了回收效率。基于这一系统,Vannatta等[11]将毛细管电泳与质谱级联,用于分选后的进一步分析。Minarik等[12]选择了另一个角度,自制了一个12通道毛细管电泳系统,并设计了一个巧妙的程控移动回收平台,通过优化收集孔板的孔断面形状以及控制移动速度和模式解决了收集问题(图1d)。其分离与收集过程中不需要中断分离电场,因此减少了毛细管中片段的扩散。随着收集平台的移动,DNA片段泳动出毛细管直接进入不同的收集板孔。但是,如果重叠片段较多,这样的设计将会大大增加后续的工作量,而且由于毛细管在收集孔板上的拖行必定带来交叉污染。
液相色谱技术为有机分子、DNA、肽和蛋白质的分离和分选提供了一个很好的自动化平台。2008年,Lim等[13]用变性高效液相色谱(DHPLC)实现了异源双链核酸的分选。但是由于精度不够,不得不采用了两次分选和两次PCR。液相色谱得到的分选量比较大,但是要消耗昂贵的柱子。
图1 基于毛细管电泳的分选方法:(a) 将ABI 310的毛细管末端单元改造,分选精度达到3 bp [7]; (b) 采用两个光纤传感器实现对快速移动的DNA片段的定位[8]; (c) 装置设计了一个石英块接口与16根毛细管连接以提高通量[9];(d) 在毛细管末端设计了一个收集DNA片段的可移动平台,实现了连续采样,以保证不丢失目标物[12]
上述方法实现了自动化操作,比传统手动人工电泳/切胶提高了效率和准确度,但是仍然存在不足,这主要是因为受到本身毛细管一维(1D)分离系统的限制,不能做到目标分选的实时性检测与处理,一些环节增大了死体积,导致了交叉污染。
4 电泳分选芯片
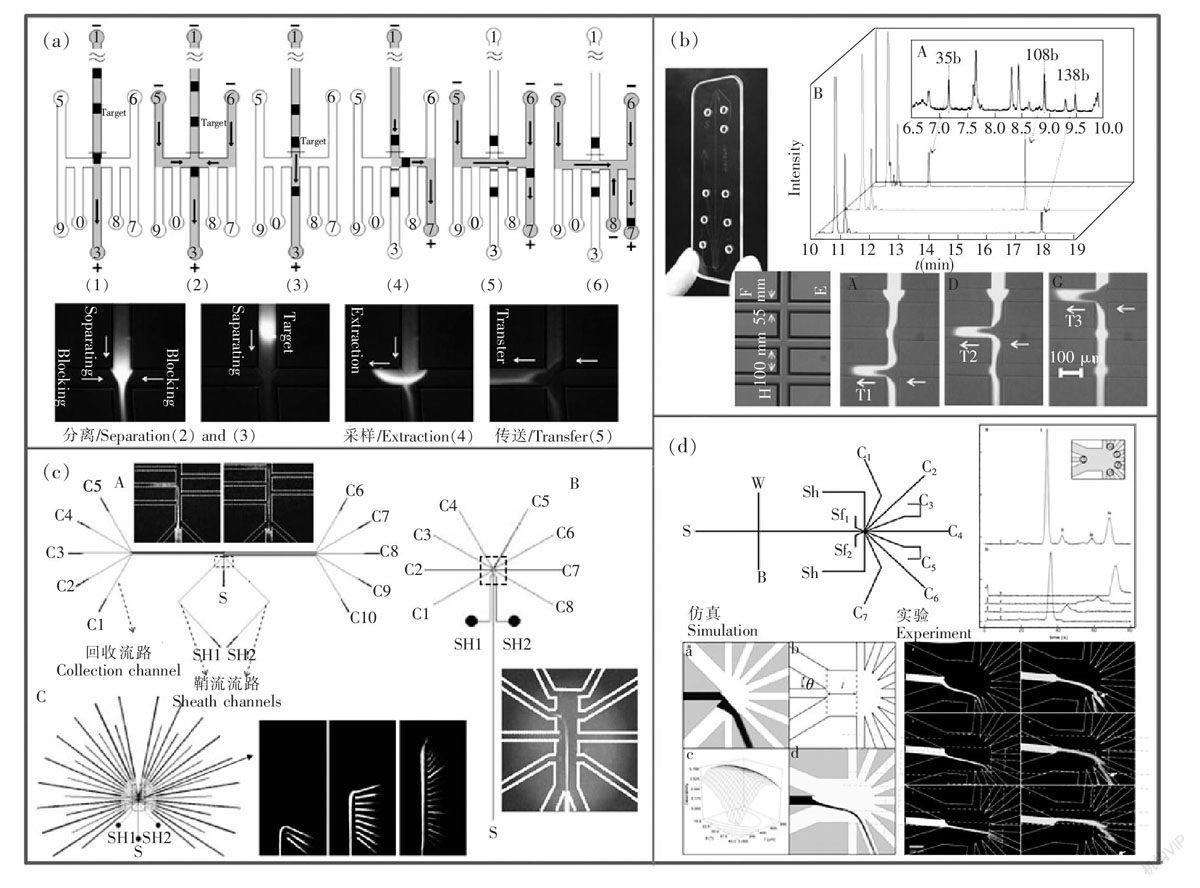
4.1 早期电泳分选芯片
随着微机电技术(MEMS)的迅速发展,生物芯片的出现成为了近年来高新技术领域中极具时代特征的重大进展,是物理学、微电子学与分子生物学综合交叉形成的高新技术。而近10年发展起来的电泳芯片已经成为第二代生物芯片/微流体芯片(微阵列芯片为第一代生物芯片)的主要成员之一。借助微泵、微阀和微混合器等集成元件的设计、加工和应用,微流控芯片越来越完善[14~16]。利用微流控电泳芯片不仅可以进行DNA长度分析,甚至可以进行序列分析和基因分型等研究[17~21]。2001年,Footz等[22]首次利用电泳芯片尝试了分离去除PCR产物中的引物和二聚体(图2a)。因为通常引物的长度相对特定产物很短,所以研究人员利用十字电泳的样品加载流路首先分离出引物,而只让产物进入分离流路部分;或者再次在分支流路加反向电压让引物返回,而只剩下移动缓慢的特定产物在交叉点附近,但是对于不同的产物,需要多次测试样品加载时间对分离的影响。Effenhauser等[23,24]最早提出专用分选微流控电泳芯片(图2b,1995年),其设计特点是在分离通道末端增加了一个垂直的T型回收微流路,使目标片段和前后的非目标片段分时进入两侧微流路。实验做了成功的分选,但是,因为目标片段要等到所有片段都通过T型交叉点后才能迁移至目标收集池,所以效率较低,而且在抑制非目标片段污染方面没有进一步讨论。Khandurina等[25]采用十字结构替换了T型结构,逐一分选了101, 187, 299, 427和567 bp长的PCR产物,该研究的最大突破是首次在分选目标前采用了鞘流,并在分选目标时拉后分离中的其它片段,以实现非目标抑制(图2c,2002年)。但是,该研究采用的分选模式共用了一段回收流路和回收池,这既增加了污染的可能性,又降低了效率。Khandurina等也发现了这个问题,因此在论文最后提出了一个多目标捕获的设想,而实际上这个设想的实现还需要进一步的污染抑制设计。从另外的角度,为了增大样品片段峰间距,减少由于片段间重叠造成的交叉污染,Khandurina等[26]将毛细管接到十字结构芯片上以提高分离精度(图2d),但是毛细管与芯片微流路的对准是非常困难的,同时也形成了一定的死体积,这一探索也因此没有被后来的研究者们采用。
图2 早期用于DNA片段分选的毛细管电泳芯片:(a)利用电泳芯片的十字进样结构分离去除PCR产物中的引物和二聚体[22];(b)最早的采用了T型回收单元的电泳分选芯片[24];(c)分离末端采用十字结构的分选芯片[25];(d)将毛细管和分选芯片相结合以提高分离精度从而抑制片段间交叉污染[26]
4.2 非目标片段抑制方法
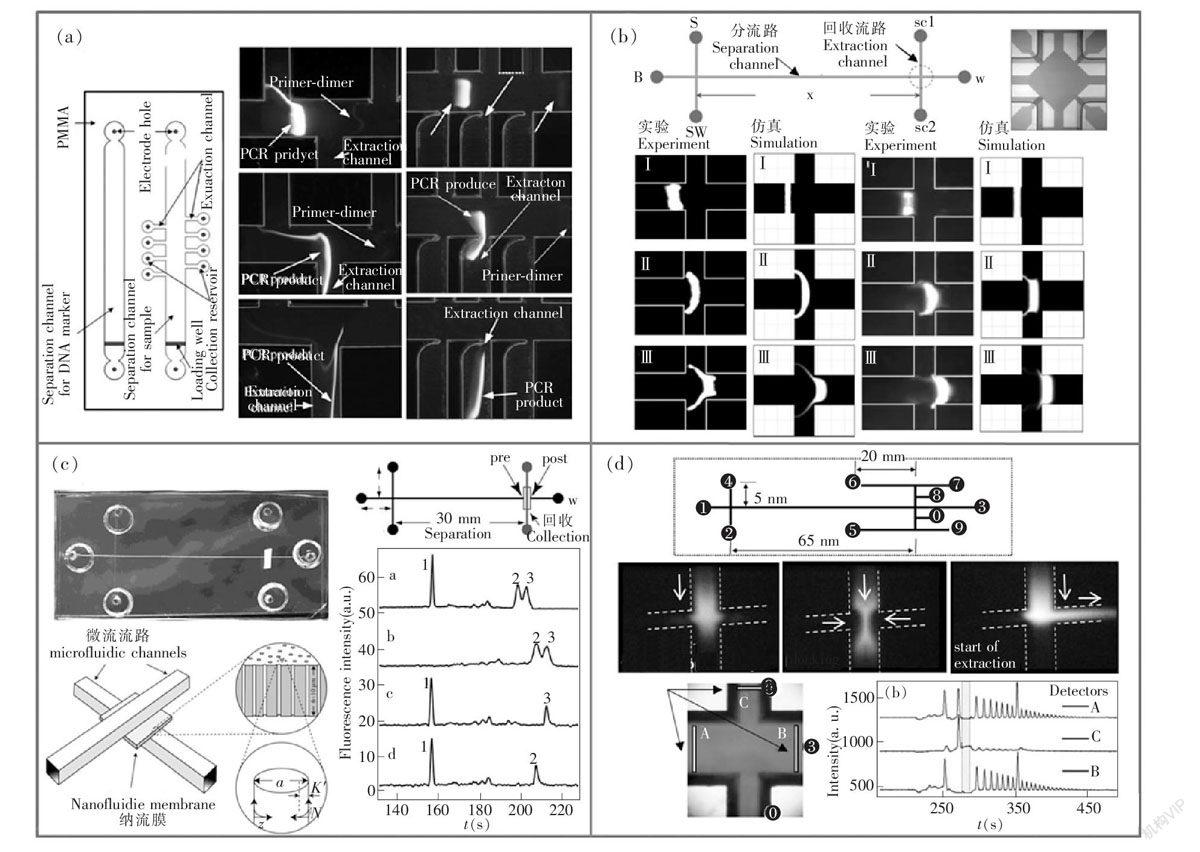
随着分选芯片研发的不断深入,研究人员意识到交叉污染抑制的重要性,因为这已经影响到分选芯片的实际应用效果。上海微系统与信息技术研究所的李刚等[27]采用垂直于分离流路配置多条平行回收流路的方法,尝试了回收流路的错位与直对布局,并优化了回收流路口形状,从而较好地抑制了片段通过垂直流路后的变形,并因此增加了回收率(图3a)。2003年,密执安大学Lin等[28]采用了类似Khandurina的简单的末端十字结构,但是改变了分选工作模式,定义的回收流路垂直于分离流路,而且在芯片上集成了分离用电极,并同时在十字流路内内置了微电极,用于抑制非目标片段,以及捕捉目标片段并将其迁移至回收流路内。Lin等[29]在2005年进一步优化了内置电极的结构,从而使非目标片段通过垂直流路时的拖尾现象得到很好的抑制(图3b)。上述几个研究都采用了仿真软件模拟了回收结构处的电场分布,这对优化设计起到了极大作用。但是,这些研究的结果仍有一定的局限,最主要的问题是分离介质采用了交联凝胶,所以片段迁移慢, 分选效率低,这些结构在非交联介质中的作用需要进一步验证。
上述研究表明,导致非目标片段通过垂直回收流路的变形和拖尾的原因,来自电场分布的变化以及分子扩散。因此减小回收流路的宽度是一个非常简单有效的办法,但是只要这个流路存在,影响就不会消失。Sweedler研究组与Beckman公司合作[30,31],在分离末端的十字结构上做了改进,如图3c所示,芯片将分离的十字电泳微流路与回收流路分置在上下两层基板,在二者的交叉点通过一层6~10
SymbolmA@ m厚的200 nm孔径聚碳酸酯纳米滤膜连接。这样的结构与鞘流控制电压配合有望做到污染最小化,但是纳米通道及其附近的电场分布,以及膜抗污染性能测试需要进一步讨论。
对比上述复杂的内置微电极和微流路结构可以发现,鞘流是最简单有效的方法,而且该方法在流式细胞仪和十字电泳进样中都有应用[32~34]。图3d给出了采用和不采用鞘流时,非目标片段进入垂直回收流路的荧光检测结果,并通过在分选交叉点前后以及两侧设置多个荧光检测点,记录了目标片段分选时各点的荧光变化,清楚地反映了目标片段捕捉的效果[35]。在我们的进一步研究中,通过对回收后的样品进行PCR扩增以及分离分析,确认了鞘流有无时,非目标片段对目标片段污染的定量分析[36]。
图3 采用不同的微结构减少DNA片段在回收流路前的扩散与交叉污染: (a) 特殊的结构设计以保证最大量的采样,减少样品损失,并控制片段变形[27]; (b) 两个十字交叉结构,一个用于进样,另一个用于分选。在分选通道附近设计了微电极结构用于控制DNA片段流经时的形状,减小片段变形[29]; (c) 采样通过垂直的具有纳米通道的薄膜,最大程度的减少了片段经过时变形[30]; (d) 通过控制回收流路两侧的偏压,形成鞘流以阻挡非目标物对回收流路的污染。在分离流路前后与采样流路入口设置荧光信号采样,右下角插图显示20bp 片段从10bp dsDNA ladder 中成功分选,分选后在分离流路中已经看不到该片段[35]
4.3 高通量分选
提高通量是微流控芯片研发最重要的目标。随着材料和微加工技术的发展,毛细管电泳分离芯片的集成度越来越高,其高通量实现方式可分为并行和串行。并行分离通道已经从4通道[37]、10通道[38,39],迅速发展为12通道[40]。在串行方面,Agilent 2100系统一次上样最多达到12个,然后依次分离;Caliper公司研发的GX系列则借助芯片上的连续提取毛细管,将连续通量扩展为96或者384,不仅解决了分离介质更换,还延长了芯片的使用寿命,降低了消耗成本;此外,光学单元的成本也低于并行检测系统;但是,串行效率不如并行高。需要指出的是,上述高通量分离芯片的研发经验并不完全适用于分选。
高通量分选,即指多目标的分离回收。随着通量的增加,不仅需要在芯片上增加大量流路和样品池,有时甚至需要在芯片上集成大量微阀和微泵单元,所以芯片集成度随之增加,这导致芯片的设计越来越困难,外围检测控制单元也越来越复杂,系统成本也大幅增加。更重要的是,高通量下样品间的交叉污染问题也越来越难避免。这些因素都是分离芯片研究未遇到的问题。
目前,最经济有效的方式是通过一次分离,相继顺序回收被分离的多个目标片段。如图4a,本研究组在鞘流流路与分离流路相连接的回收流路上布置了多个回收流路分支,并将激光聚焦在回收流路口,在检测到目标样品后,通过切换电场操控目标片段进入对应的回收流路[35]。但是,不同于单一片段分选,多目标片段分选芯片有多个回收流路分支,目标片段进入对应的分支之前会对其它分支产生污染,因此,简单的鞘流已经不能对所有的回收分支流路起到保护作用。为此,我们在目标片段进入对应样品回收池后,增加了对被污染分支流路的冲洗操作,即将进入“错误”流路的片段移出并送至对应目标样品池。对于多目标分选,回收的次序是需要严格设计的,以图4a为例,目标片段进入的次序是先外侧再内侧,即7980。但是,这样的设计操作略显繁琐,而且提取回收样品时容易出错,为后续工作增
图4 多目标片段分选芯片:(a)共用回收流路的4目标分选芯片,给出了清晰的分选流程和片段移动方向[35];(b)隔离型3目标分选芯片及回收后电泳图谱[41];(c)采用树状、星形和36通道的高通量分选芯片[42];(d)以实现连续回收为目的,采用圆周分布型的多目标回收结构设计,样品经过3.5 cm的分离后,在鞘流的作用下连续扫过回收流路[44]
加了隐患;因为与分离流路垂直的回收流路成为了共用流路,所以也成为了交叉污染的隐患。为此,在进一步的研究中,将回收流路全部平行隔离开,以减少彼此共用部分(图4b)[41]。最上面的回收流路也是鞘流流路,荧光实验证明
优化后的鞘流可以保护下面两个流路,自下而上依次回收的片段长度为35, 108和138 bp,其纯度为99.3%, 97.9%和94.7%。虽然纯度都很高,在后续的测序当中也没有发现异常,但是在重复实验中发现,纯度呈下降趋势。深入分析发现,主要原因是分选前一个目标时,主分离流路中的样品由于电场作用停止而增大了扩散,同时DNA分子不再是拉直状态,分离精度因此下降;另外DNA分子与管壁的吸附也需要考虑。为了解决上述问题,Harrrison课题组[42]加宽了鞘流后的流路,减少了样品与回收流路的接触,以达到降低污染的目的(图4c)。该研究最大推出了36通道分选芯片,但是作者未对全部通道的回收样品做对应的纯度分析,如果该设计能够保证高通量下的低污染将非常有意义。2013年,该课题组进一步研发了用于蛋白组学研究的样品分选、富集和洗脱的6通道以及8通道芯片[43]。图4d则进一步缩短了鞘流后的流路长度,并将回收流路呈圆周状布置,使电场分布更一致,同时最大程度提高了通量;设计最大优点是,在分选过程中,主分离流路电场不需要切断,片段被连续依次送入回收流路[44]。这个设计原理与前面图2d的机械连续运动方式回收机构相似,优势是采用了快速的电扫描方式,不足是通量不足以应对尺寸接近或者重叠片段分选。
虽然上述设计具有了很大改进,但是在复杂目标分选过程中,尤其是重叠片段分选问题仍然没有解决。2009年。本研究组实现了单碱基差的3个部分重叠的单一目标片段分选[45],相继回收的3个片段的纯度均超过90%,部分达到99%,至此,将分选精度推到了极限。但是分离时间超过40 min,而且仅能做单一目标分选。本研究组根据采样定理设计了一款新的芯片,采用10条间距为100 μm的回收流路构成采样区,在样品片段进入采样区后,10条回收流路同时回收采样区内的样品(图5a)。采用新芯片仅用10 min就实现了单碱基差重叠片段样品的分选[46];而且一次采样成功回收了相邻的两个片段[47]。这种方法改变了以往断片回收以分离精度为必要条件的限制,大大缩短了分离时间,提高了效率。
图5 几种新颖的多目标片段分选芯片:(A)运用采样定理的同时空间分选芯片[47];(B)具有棱镜式分离性能的DNA大分子分选结构[48];(C)可分离两种混合物的二维自由流电泳芯片[49]
以往的DNA片段回收量少,所以必须通过PCR对回收样品进行浓度扩增,这无疑增加了后续工作量。2002年,Huang等[48]以2 μm圆柱矩阵为分离介质的双向电泳芯片,新颖结构的作用如同光散射棱镜,将长度为61, 114, 158和209 kb的大分子DNA片段呈不同角度分离开,并在末端实现连续回收(图5b)。同样采用连续流分离回收方式,日本NEC公司与BABA研究组合作,将分离柱直径从2 μm降到了200 nm以下,柱间缝隙仅约为32 nm宽,以此将回转直径57 nm的300 bp DNA片段和回转直径22 nm的100 bp DNA片段通过不同通道分别回收[49]。纳米柱或者纳米壁形成的矩阵在分离过程中可以拉长核酸分子[50,51],这对于核酸大分子分离和回收具有极大潜力,甚至有望推动实现DNA单分子测序。2009年,Zalewski等[52]设计了另一款二维自由流电泳芯片,通过调控3个输入和5个输出端的交流电场,以连续流的方式将两个混合的样品送入不同的输出端(图5c)。这两个研究的突出优点是解决了DNA大分子的连续多目标分选,而且连续分选可以获得大量富集,而无需以往方法的PCR浓度扩增。
4.4 商用分选仪器
2010年,美国Caliper LifeSciences公司推出了Labchip XT自动化核酸回收仪器,使用的是4或5通道一次性芯片,工作流程与前面的研究团队也基本一致。通过芯片采用的试剂盒和芯片设计可以看出,Caliper公司并没有采用芯片电泳的经典十字进样设计,分离介质与分离原理与凝胶电泳类似,这说明该产品更强调回收量,以此减少PCR扩增带来的成本增加和问题复杂化。但是,这也同时继承了凝胶电泳的缺点,精度低仅为5 b/bp;分离回收时间长,快的需要30 min完成,有的甚至长达50~100 min。
值得提出的是,Labchip XT所提出的非液相接触式电极,避免了前后两块芯片之间通过电极产生的交叉污染。其解决办法是在芯片上再内置一套金属电极,内置电极与缓冲液接触。另外值得称赞的是,产品软件设计严密,确保了大部分分选的重复性,节省了人力,提高了效率。从其设计可以看出,单通道原则上一次只能回收一个目标片段,虽然系统设计提供了“Extraction and Pause”功能,在手动提取第一个目标片段后,可以继续执行下一个目标片段的分选,但是之间要对回收池进行手动清洗,以避免样品间的交叉污染。这大大限制了效率的提高。
上述仪器虽然仍存在很多不足,但是自动化分选仍使很多新一代测序用户接受了这一产品,实际使用表明,使用自动化样品制备平台,配合核酸电泳分离分析设备,能够将全外显子捕获及提高相应测序文库构建的效率,在确保最终测序数据质量的前提下,从原本人工操作时的每周每人12样提升至每周每人200 样以上。但是,对复杂的具有重叠片段的分选,目前市场上的仪器仍无法进行高精度高重复性的分选。另外需要指出的是,与成千上万的目标片段相比,仅4或5个通道的分选还是显得不足,而且样品提取后没有纯度分析,自动化的优势因此逊色不少。
5 结 语
综上所述,虽然分选方法在不断改善,精度在不断提高,但是快速实现高准确度、高通量和高纯度的自动化分选和各种复杂目标峰的分选与定量,是目前科研人员面对的新挑战,主要包括:(1)高电场下快速移动片段的准确捕捉 细小的流路使更高电场的使用成为可能,因此片段的迁移速度更快,分选过程的完成将得到速度提升。因此需要新的方法实现准确的捕捉,而又避免相邻片段的污染。(2)重叠片段的准确分选方法 即使良好的分离也不能完全避免重叠片段的出现,而良好的分离往往需要更长的分离距离和时间。因此研究新的分选方法实现短距离分离、在重叠状态下实现高纯度分选非常重要。(3)实用化高通量分选方法 这是提高处理能力和效率,应对实际需求必须面对的问题。需要解决的主要是芯片设计和检测方法。(4)建立分选原位分析于一体的自动化体系 单独的自动化分选仪器可以解决上述瓶颈问题,但在多数情况下,分选后的样品仍需扩增富集和纯度分析,两个流程之间不可避免的手工操作牺牲掉了刚刚获得的优势,同时增加了样品被污染的几率。所以实现完整的自动化流程非常重要。
该领域的研究属于生物芯片发展前沿,同时我国在以生物工程、分子生物学等为代表的生命科学各个领域,对各种对象的微量分选制备存在迫切需求。因此,对国内的科研工作者而言,以微流控芯片为载体,充分利用微流控芯片设计在二维空间的延展,发挥它在集成化、最小化、低样品试剂消耗、高效率、实时检测等方面的优势,对建立具有国际竞争力的高端筛选仪器体系和品牌具有积极作用。
References
1 Ding L, Stilwell J, Zhang T, Elboudwarej O, Jiang H, Selegue J P, Cooke P A, Gray J W, Chen F F. Nano Lett., 2005, 5(12): 2448-2464
2 Heidi L. Nature, 2008, doi:10.1038 /news. 2008.439
3 Collins F S, Morgan M, Patrinos A. Science, 2003, 300(5617): 286-290
4 Fukumura R, Takahashi H, Saito T, Tsutsumi Y, Fujimori A, Sato S, Tatsumi K, Araki R, Abe M. Nucleic Acids Res., 2003, 31(16): e94-e94
5 Kostal V, Katzenmeyer J, Arriaga E A. Anal. Chem., 2008, 80(12): 4533-4550
6 Kodzińska E, Buszewski B. Anal. Chem., 2009, 81(1): 8-15
7 Lin M, Rich R G, Shipley R F, Hafez M J. J. Mol. Diagn., 2007, 9(5): 598-603
8 Minarik M, Foret F, karger B L. Electrophoresis, 2000, 21(1), 247-254
9 Irie T, Oshida T, Hasegawa H, Matsuoka Y, Li T, Oya Y, Tanaka T, Tsujimoto G, Kambara H. Electrophoresis, 2000, 21(2): 367-374
10 Huge B J, Flaherty R J, Dada OO, Dovichi N J. Talanta, 2014, 130(1): 288-293
11 Vannatta M W, Whitmore C D, Dovichi N J. Electrophoresis, 2009, 30(23): 4071-4074
12 Minarik M, Kleparnik K, Gilar M, Foret F, Miller A W, Sosic Z, Karger B L. Electrophoresis, 2002, 23(1): 35-42
13 Lim K S, Naviaux R K, Wong S, Haas R H. J. Mol. Diagn., 2008, 10(1): 102-108
14 Mitchell P. Nat. Biotechnol., 2001, 19: 717-721
15 Auroux P A, Iossifidis D, Reyes D R, Manz A. Anal. Chem., 2002, 74(12): 2637-2652
16 Reyes D R, Iossifidis D, Auroux P A, Manz A. Anal. Chem., 2002, 74(12): 2623-2636
17 Harrison D J, Fluri K, Seiler K, Fan Z, Effenhauser C S, Manz A. Science, 1993, 261(5123): 895-897
18 Woolley A T, Mathies R A. Anal. Chem., 1995, 67(20): 3676-3680
19 Shi Y, Anderson R C. Electrophoresis, 2003, 24(1920): 3371-3377
20 Yi LL, Xu X Q, Lin X X, Li H F, Ma Y, Lin J M. Analyst, 2014, 139(13): 3330-3335
21 Deng Y, Yi LL, Lin X X, Lin L, Li H F, Lin J M. Talanta, 2015, 144: 136-144
22 Footz T, Wunsam S, Kulak S, Crabtree H J, Glerum D M, Backhouse C J. Electrophoresis, 2001, 22(18): 3868-3875
23 Effenhauser C S, Manz A, Widmer H M. Anal. Chem., 1993, 65(19): 2637-2642
24 Effenhauser C S, Manz A, Widmer H M. Anal. Chem., 1995, 67(13): 2284-2287
25 Khandurina J, Chovan T, Guttman A. Anal. Chem., 2002, 74(7): 1737-1740
26 Khandurina J, Guttman A. J. Chromatogr. A, 2002, 979(12): 105-113
27 Li G, Ran R, Zhao J, Xu Y. Electrophoresis, 2007, 28(24): 4661-4667
28 Lin R, Burke D T, Bruns M A. J. Chromatogr. A, 2003, 1010(2): 255-268
29 Lin R, Burke D T, Bruns M A. Anal. Chem., 2005, 77(14): 4338-4347
30 Kuo T, Cannon D M, Chen Y, Tulock J J, Shannon M A, Sweedler J V, Bohn P W. Anal. Chem., 2003, 75(8): 1861-1867
31 Tulock J J, Shannon M A, Bohn P W, Sweedler J V. Anal. Chem., 2004, 76(21): 6419-6425
32 Bharadwaj R, Santiago J G, Mohammadi B. Electrophoresis, 2002, 23(16): 2729-2744
33 Dittrich P S, Schwille P. Anal. Chem., 2003, 75(21): 5767-5774
34 Fu L M, Yang R J, Lee G B. Anal. Chem., 2003, 75(8): 1905-1910
35 Sun K, Li Z, Ueno K, Juodkazis S, Noji S, Misawa H. Electrophoresis, 2007, 28(10): 1572-1578
36 Sun K, Suzuki N, Li Z Y, Araki R, Ueno K, Juodkazis S, Abe M, Noji S, Misawa H. Electrophoresis, 2008, 29(19): 3959-3963
37 Shen Z, Liu X J, Long Z C, Liu D Y, Ye N N, Qin J H, Dai Z P, Lin B C. Electrophoresis, 2006, 27(56): 1084-1092
38 Dang F, Shinohara S,Tabata O, Yamaoka Y, Kurokawa M, Shinohara Y, Ishikawa M, Baba Y. Lab Chip, 2005, 5(4): 472-478
39 Dang F, Tabata O, Kurokawa M, Ewis A A, Zhang L, Yamaoka Y, Shinohara S, Shinohara Y, Ishikawa M, Baba Y. Anal. Chem., 2005, 77(7): 2140-2146
40 Inoue A, Ito T, Makino K, Hosokawa K, Maeda M. Anal. Chem., 2007, 79(5): 2168-2173
41 Li Z Y, Sun K, Sunayama M, Araki R, Ueno K, Abe M, Misawa H. J. Chromatogr. A, 2011, 1218(7): 997-1003
42 Wang Z, Justine T, Jemere A B, Harrison D J. Electrophoresis, 2010, 31(15): 2575-2583
43 Hua Y, Jemere A B, Dragoljic J, Harrison D J. Lab Chip, 2013, 13(13): 2651-2659
44 Baker C A, Roper M G. J. Chromatogr. A, 2010, 1217(28): 4743-4748
45 Sun K, Suzuki N, Li Z Y, Araki R, Ueno K, Juodkazis S, Abe M, Noji S, Misawa H. Electrophoresis, 2009, 30(24): 4277-4284
46 Li Z Y, Sun K, Sunayama M, Araki R, Ueno K, Abe M, Misawa H. Electrophoresis, 2011, 32(23): 3392-3398
47 Sun K, Li Z Y, You S J, Zhang X Y, Ren N Q. Microfluid. Nanofluid., 2015, 19(2): 291-298
48 Huang R L, Jonas T O, Jessica K J, James S C, Robert A H, Cox E C. Nat. Biotech., 2002, 20: 1048-1051
49 Hattori W, Someya H, Baba M, Kawaura H. J. Chromatogr. A, 2004, 1051(12): 141-146
50 Yasui T, Kaji N, Ogawa R, Hashioka S, Tokeshi M, Horiike Y, Baba Y. Anal. Chem., 2011, 83(17): 6635-6640
51 Kazemlou S, Nazemifard N. Microfluid. Nanofluid., 2014, 17(6): 993-1002
52 Zalewski D R, Gardeniers H J. Electrophoresis, 2009, 30(24): 4187-4194
Advance in Microfluidic Devices for Fractionation of DNA Fragments
LI ZheYu, SUN Kai*, ZHANG XiaoYan, LIU ShaoQin, JIANG Lei, REN NanQi
(State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150090, China)
Abstract Current next generation sequencing works faster and the price is coming down. However, the workflows have numerous manual processes, which contribute to bottleneck and process inefficiency. One of the most time consuming steps is electrophoretic gelbased fractionation of a large number of fragments of interest from the library generation process. In recent years, several instruments were first introduced into the market, and then were tested soon by many famous DNA sequencing centers and platforms. This paper introduces the development of DNA fractionation techniques including capillary electrophoresis and microfluidic devices. Moreover, our argument raises the bottleneck in fractionating DNA fragments on the chips. Finally, we provide insights into the challenges of DNA fractionation and perspectives.
Keywords Capillary electrophoresis; Microfluidic chip; Fractionation; DNA sequencing; Gene expression; Review
(Received 23 July 2015; accepted 28 September 2015)
This work was supported by the State Key Laboratory of Urban Water Resource and Environment (No. 2011TS02)
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
科技信息·学术版(2022年8期)2022-02-25
华东师范大学学报(自然科学版)(2019年5期)2019-11-11
华东师范大学学报(自然科学版)(2019年5期)2019-11-11
华东师范大学学报(自然科学版)(2019年5期)2019-11-11
智富时代(2018年7期)2018-09-03
智富时代(2018年7期)2018-09-03
江苏农业科学(2016年8期)2017-02-15
中国实用医药(2016年30期)2016-12-28
科技视界(2016年16期)2016-06-29
