
藿香正气合剂的HPLC指纹图谱研究
2016-11-18 00:34:28林雀跃张荣林
中国民族民间医药 2016年20期
林雀跃 张荣林 罗 轶 张 慧
广西壮族自治区食品药品检验所,广西 南宁 530021
藿香正气合剂的HPLC指纹图谱研究
林雀跃 张荣林 罗 轶 张 慧
广西壮族自治区食品药品检验所,广西 南宁 530021
目的:建立藿香正气合剂的HPLC指纹图谱。方法:样品直接进样,以橙皮苷为参照峰,通过紫外检测器(波长280nm)对藿香正气合剂进行HPLC指纹图谱分析。结果:对20批藿香正气合剂供试品进行检测,建立了该药品的HPLC指纹图谱并标示了15个共有指纹峰。各共有峰相对保留时间RSD均在1.00%以内。结论:该方法准确、重现性好,为藿香正气合剂的进一步质量标准化研究和控制提供依据。
藿香正气合剂;指纹图谱;HPLC
藿香正气合剂由广藿香、紫苏叶、白芷、白术、陈皮、茯苓、桔梗、甘草、大腹皮、姜厚朴、姜半夏等13味药材组成,具有解表化湿、理气和中的功效,临床用于脾胃虚寒而外感风寒、胸膈闷满。收载于《中华人民共和国卫生部药品标准中药成方制剂》第7册中,原标准没有含量测定项[1]。为更好的控制该药品的质量,张志平等[2]采用薄层扫描法测定了藿香正气合剂中百秋李醇的含量;也有企业用高效液相色谱法测定橙皮苷含量作为其质控指标,但关于藿香正气合剂指纹图谱方面的研究未见报道。中药指纹图谱是一种实现对中药多组分,多指标分析的有效方法,该技术具有整体性、特征性、模糊性和稳定性的特征,可从整体上把握中药的质量[3]。从藿香正气类制剂指纹图谱研究的进展看[4-7],藿香正气水的指纹图谱研究较多,但藿香正气水以挥发油类成分为主,而藿香正气合剂则以水溶性成分为主,因此其指纹图谱的研究思路和方法与藿香正气水截然不同。研究通过实验摸索,采用样品直接进样的方法对藿香正气合剂进行HPLC指纹图谱分析,并在方法学考察的基础上,建立了藿香正气合剂的指纹图谱,同时,以保留时间为指标,通过对比藿香正气合剂、药材样品提取液及缺某味药材阴性样品HPLC指纹图谱,对15个共有峰进行归属指认。该法具有操作简单、准确、重现性好等特点,为其质量控制提供了依据。
1 仪器与试剂
1.1 仪器 Agilent1200型液相色谱仪(真空脱气装置、四元泵、自动进样器和DAD检测器、UWD紫外检测器);MILLI-PROA纯水处理器。
1.2 试剂 橙皮苷(批号:110721-201115,中国食品药品检定研究院);藿香正气合剂20批(无糖型10批的批号:131215、131216、131217、131220、131221、131222、131224、131225、131226、131227,样品序号为S1~S10;有糖型10批,样品序号为S11~S20)、藿香正气合剂阴性样品13批(缺广藿香阴性、缺紫苏叶阴性、缺白芷阴性、缺白术阴性、缺陈皮阴性、缺茯苓阴性、缺桔梗阴性、缺甘草阴性、缺大腹皮阴性、缺厚朴阴性、缺半夏阴性、缺生姜阴性、缺大枣阴性)均由广西邦琪药业集团有限公司提供;单味药材提取液13批(广藿香提取液、紫苏叶提取液、白芷提取液、白术提取液、陈皮提取液、茯苓提取液、桔梗提取液、甘草提取液、大腹皮提取液、厚朴提取液、半夏提取液、生姜提取液、大枣提取液)按藿香正气合剂制法单独提取。乙腈为色谱纯,水为超纯水,其余试剂均为分析纯。
2 方法
2.1 色谱条件 色谱柱:CAPCELLPAK MGⅡ C18(4.6mm×250mm,5μm);流动相:乙腈(A)-0.1%磷酸溶液(B)梯度洗脱程序(见表1);流速:1.0mL/min;检测波长:280nm;柱温:35℃;进样量:10μL。理论板数按橙皮苷峰计算应不低于5000。

表1 流动相洗脱程序
2.2 供试品溶液的制备 取样品溶液过0.45μm微孔滤膜,即得。
2.3 测定方法 精密吸取供试品溶液10μL,注入液相色谱仪,测定并记录液相色谱图,分析该色谱图60min内的色谱峰信息,20批样品均出现15个明显的液相色谱峰,将其确定为共有峰,计算各共有峰的相对保留时间和相对峰面积。
2.4 精密度实验 取藿香正气合剂,连续测定6次,以橙皮苷峰作为参照峰(S峰),考察各共有峰的相对保留时间(Tr=Ti/Ts)、相对峰面积(Ar=Ai/As)的一致性。结果表明:各共有峰相对保留时间RSD为0.04%~0.39%;相对峰面积RSD为0.56%~2.28%。
2.5 稳定性实验 取藿香正气合剂,分别在0、8、16、24、32、40h检测,结果在40h内,各共有峰相对保留时间RSD为0.07%~1.13%,相对峰面积RSD为0.38%~2.28%,表明样品溶液打开包装后在40h内测定稳定。
2.6 重复性实验 取藿香正气合剂6份,按“2.1”项实验条件分析,结果6份样品各共有峰相对保留时间RSD为0.02%~0.17%,相对峰面积的RSD为0.47%~2.00%。表明方法的重复性很好。
2.7 耐用性实验 考察3个品牌色谱柱[CAPCELLPAK MGⅡC18、Kromasil 100-5 C18、Inertsil ODS-3 C18(4.6mm×250mm,5μm)],结果藿香正气合剂各共有峰相对保留时间的RSD为0.57%~5.61%,相对峰面积的RSD为0.23%~5.72%。表明采用相同仪器不同色谱柱,对少数共有峰的出峰时间及峰面积有影响,但影响不大。
3 结果与讨论
3.1 HPLC指纹图谱的确立及共有峰归属 在“2.1”项的实验条件下,对20批藿香正气合剂进行分析,确定了15个共有峰(见图1),建立了藿香正气合剂HPLC指纹图谱。以橙皮苷峰为参照峰,其保留时间和峰面积分别定为1.00,计算各共有峰的相对保留时间和相对峰面积(见表2、表3)。从数据上可以看出:不同批次样品同一色谱共有峰的相对面积存在差异,这可能与各单味药材的来源、采集时间和制药工艺的稳定性有关。

表2 20批藿香正气合剂HPLC各共有峰相对保留时间表

序号峰1峰2峰3峰4峰5峰6峰7峰8峰9峰10峰11峰12峰13峰14峰15S10.19520.35250.38030.40140.46240.48410.51120.58850.79190.86330.90090.93371.00001.20241.2408S20.19590.35380.38140.40270.46380.48530.51260.58920.79320.86450.90150.93441.00001.20341.2425S30.19480.35270.38210.40210.46370.48480.51250.58850.79330.86240.90180.93531.00001.20401.2436S40.19520.35230.38040.40120.46210.48440.51130.58810.79210.86180.90070.93411.00001.20211.2402S50.19560.35280.38110.40200.46250.48410.51130.58810.79170.86210.90110.93421.00001.20331.2415S60.19510.35210.38050.40110.46140.48440.51150.58840.79180.86090.90050.93401.00001.20201.2396S70.19460.35140.37960.40000.46090.48400.51130.58740.79070.86050.90070.93421.00001.20261.2410S80.19590.35840.37820.40750.47050.48450.51300.59130.79820.86700.90030.93321.00001.19331.2398S90.19910.36490.38350.41310.47560.48910.51690.59330.79780.86750.90180.93411.00001.20061.2409S100.19490.35250.38040.40190.46320.48440.51190.58820.79310.86240.90140.93451.00001.20341.2428S110.19490.35370.38430.40460.46540.48530.51310.58950.79560.86230.90170.93521.00001.20361.2440S120.19480.35280.38240.40360.46410.48550.51310.58940.79500.86160.90200.93551.00001.20421.2439S130.19480.35270.38210.40360.46370.48530.51280.58870.79430.86090.90160.93551.00001.20451.2440S140.19480.35250.38130.40380.46410.48510.51280.58930.79490.86140.90190.93561.00001.20461.2441S150.19450.35250.38160.40380.46450.48570.51340.58970.79490.86140.90180.93551.00001.20401.2438S160.19490.35330.38360.40430.46440.48570.51370.59010.79570.86190.90210.93571.00001.20481.2448S170.19440.35250.38180.40350.46380.48480.51260.58900.79470.86050.90160.93551.00001.20451.2438S180.19460.35250.38150.40360.46430.48560.51320.58920.79460.86090.90190.93571.00001.20451.2443S190.19480.35280.38180.40410.46460.48540.51340.58920.79440.86110.90180.93551.00001.20531.2448S200.19480.35260.38130.40360.46430.48570.51350.58930.79480.86130.90210.93581.00001.20451.2442均值0.19520.35360.38150.40360.46450.48520.51280.58920.79410.86230.90150.93491.00001.20311.2427RSD/%0.520.850.370.690.700.230.250.220.250.230.060.090.000.210.14

表3 20批藿香正气合剂HPLC各共有峰相对峰面积表
对15个共有峰进行归属分析可知:峰1由白芷、白术、陈皮等9味药材贡献;峰2由广藿香、紫苏叶、厚朴等3味药材贡献;峰3由陈皮、大腹皮、厚朴等3味药材贡献;峰4由白芷、白术、甘草、厚朴等4味药材贡献;峰5由广藿香、紫苏叶、白芷、甘草、厚朴等5味药材贡献;峰6由陈皮、厚朴2味药材贡献;峰7由广藿香、甘草、厚朴、陈皮、紫苏叶等5味药材贡献;峰8由紫苏叶、厚朴2味药材贡献;峰9、峰10同由紫苏叶、甘草2味药材贡献;峰11为陈皮药材特征峰;峰12为厚朴药材特征峰;峰13为橙皮苷峰;峰14为广藿香药材特征峰;峰15为甘草药材特征峰。(见图2、表4)

表4 藿香正气合剂15个共有峰归属表
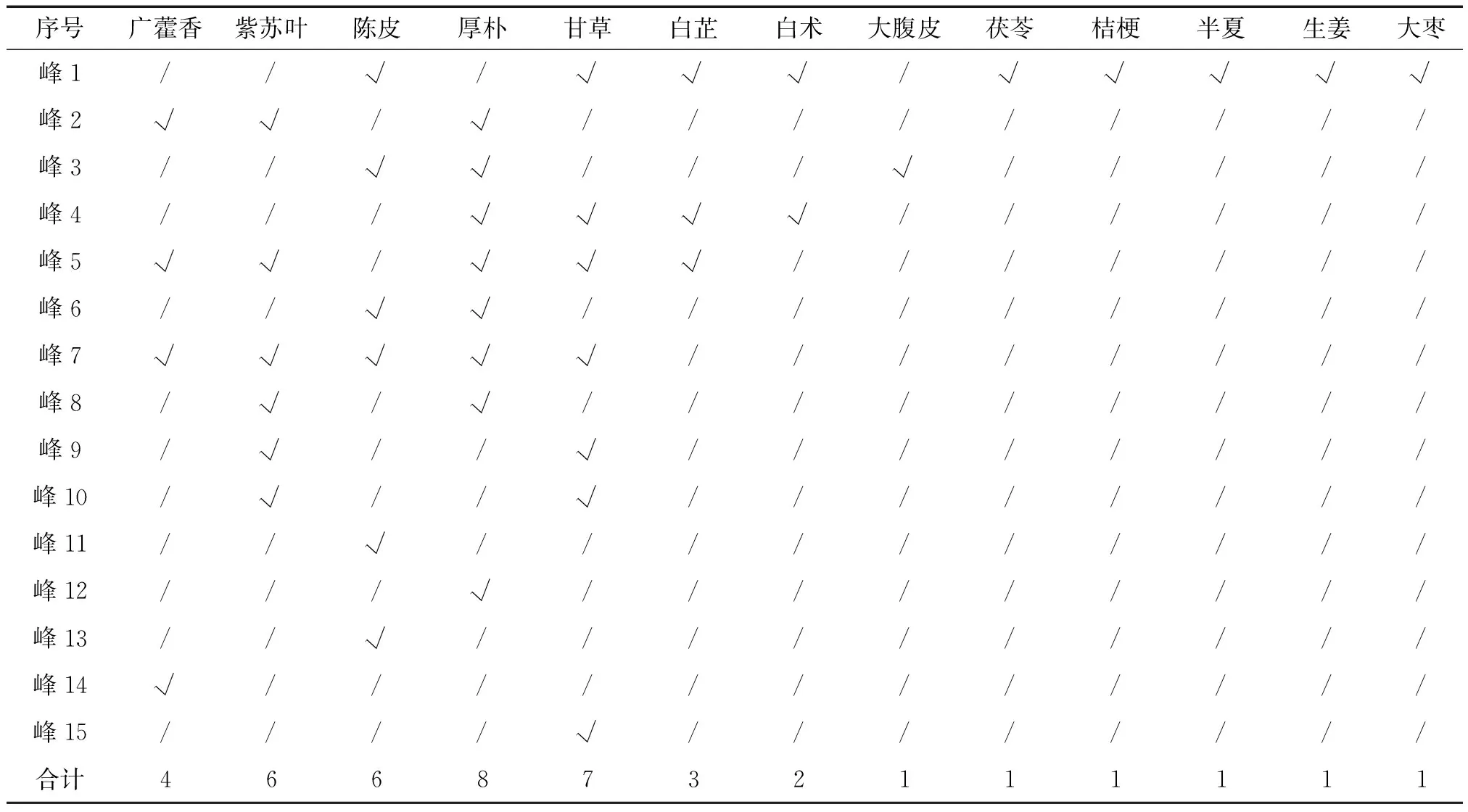
序号广藿香紫苏叶陈皮厚朴甘草白芷白术大腹皮茯苓桔梗半夏生姜大枣峰1//√/√√√/√√√√√峰2√√/√/////////峰3//√√///√/////峰4///√√√√//////峰5√√/√√√///////峰6//√√/////////峰7√√√√√////////峰8/√/√/////////峰9/√//√////////峰10/√//√////////峰11//√//////////峰12///√/////////峰13//√//////////峰14√////////////峰15////√////////合计4668732111111
3.2 实验条件的选择 在DAD检测器中查看全波长色谱图,发现样品于280nm波长下得到的色谱峰数量较多且峰面积较大,因此确定280nm作为检测波长。对比了Ⅰ:乙腈-水、Ⅱ:甲醇-0.1%磷酸、Ⅲ:乙腈-0.1%磷酸3个流动相系统,结果显示系统Ⅲ的峰对称性较好、分析时间较短。藿香正气合剂以水溶性成分为主,极性较大,故考察了Ⅰ:原液直接进样、Ⅱ:D101型大孔树脂柱富集净化、Ⅲ:聚酰胺柱富集净化3种制备方法。结果表明采用D101型大孔树脂柱及聚酰胺柱对本品进行富集纯化效果不显著,最终确定用藿香正气合剂样品直接进样。

对15个共有峰的Fi值进行排序,可知峰15>峰3>峰8>峰5>峰6>峰2>峰1>峰4>峰12>峰7>峰14>峰9>峰10>峰11>峰13。峰13、11变化差异最小,两峰均为陈皮的特征峰,表明陈皮药材的质量较稳定,且藿香正气合剂制备工艺对陈皮药材成分的提取影响较小,也验证了选用陈皮特征峰(橙皮苷)为参照峰评价藿香正气合剂质量是合理的。峰15的变化差异最大,其次为峰3、8、5、6、2。其中峰15为甘草特征峰,后五个峰均由厚朴参与贡献,表明,甘草及厚朴药材对藿香正气合剂质量的影响最大,这与每批投料的甘草、厚朴药材质量良莠不齐有关,因此,把好该两味药材进厂的质量关是关键。14号峰为君药广藿香的特征峰,各批次相对峰面积值较为稳定,可通过监控多批藿香正气合剂的14号峰相对峰面积值来确定一个数值范围作为其质控指标。
4 结论
从20批藿香正气合剂HPLC指纹图谱分析结果来看,无糖型及有糖型样品HPLC指纹图谱相似度高,表明蔗糖辅料的使用对该药品的化学成分无显著影响;采用样品直接进样分析的方法最大程度的保留了该药品中化学成分的种类,节省了样品提取的时间,操作简单;参与藿香正气合剂HPLC指纹图谱共有峰贡献排名前五的药材依次为厚朴、甘草、紫苏叶、陈皮及广藿香,表明这五味药材的质量直接影响该制剂的质量,可通过测定五味药材HPLC指纹图谱,筛选出质量稳定的优质药材进行投料,进而达到控制藿香正气合剂质量的目的。研究表明,藿香正气合剂HPLC指纹图谱能有效控制该制剂质量。
[1]中华人民共和国卫生部.中华人民共和国卫生部药品标准·中药成方制剂(第七册)[S].1993:206.
[2]张志平,戴晓弘.薄层扫描法测定藿香正气合剂中百秋李醇的含量[J].中医研究,2005,18(10):18-19.
[3]周建良,齐炼文,李萍.色谱指纹图谱在中药质量控制中的应用[J].色谱,2008,26(2):153-155.
[4]张良圣,倪永年.应用高效液相色谱法和化学计量学研究藿香正气水指纹图谱[J].南昌大学学报(理科版),2007,31(1):93-96.
[5]李红梅,茹鑫,梁悦,等.藿香正气水气相指纹图谱特性研究[J].中成药,2010,32(1):6-10.
[6]刘征辉,叶挺祥,赵洪芝,等.藿香正气水指纹图谱及模式识别对质量控制的研究[J].药物分析杂志,2012,32(1):2024-2030.
[7]阚红玉,李昂,葛丹丹,等.藿香正气滴丸的波长切换法指纹图谱研究[J].现代中药研究与实践,2014,28(2):55-57.
[8]冯毅凡,苏薇薇,吴忠.华佗再造丸气相色谱指纹特征鉴别及质量评价[J].中药材,1999,22(7):373-376.
(编辑:陶希睿)
HPLC Fingerprint of Huoxiang Zhengqi Mixture
LIN Queyue ZHANG Ronglin LUO Yi ZHANG Hui
Guangxi Institute for Food and Drug Control,Nanning 530021,China
Objective To establish the HPLC fingerprint of Huoxiang Zhengqi Mixture. Methods The constituents of Huoxiang Zhengqi Mixture were analyzed by HPLC with UV detector (wavelength 280nm) using direct sampling and hesperidin used as the
ubstance. Results HPLC fingerprint of Huoxiang Zhengqi Mixture,15 common peaks were established on the basis of system atic methodology after 20 batches of samples were tested Variation in the relative retention time of 15 identified common peaks were within 1.00% range. Conclusion The analytical method for Huoxiang Zhengqi Mixture is precise and reliable.The research would be helpful to offer an effective pattern for quality control of Huoxiang Zhengqi Mixture.
Huoxiang Zhengqi Mixture;Fingerprint;HPLC
2016-08-24
林雀跃(1983-),女,汉族,主管中药师,硕士,从事中药材及中成药质量评价。E-mail:linqueyue@163.com
R284
A
1007-8517(2016)20-0043-05
猜你喜欢
中老年保健(2022年6期)2022-11-25 13:49:39
华人时刊(2022年11期)2022-09-15 00:54:34
散文诗(青年版)(2022年4期)2022-04-25 23:52:34
家教世界(2021年34期)2022-01-13 12:04:44
基层中医药(2021年8期)2021-11-02 06:24:56
基层中医药(2021年10期)2021-06-05 07:15:26
保健与生活(2020年17期)2020-09-27 08:10:18
基层中医药(2020年12期)2020-07-22 06:35:02
中成药(2018年10期)2018-10-26 03:41:10
中成药(2018年10期)2018-10-26 03:41:02
