
渣油四组分吸附与裂化反应性能研究①
2016-11-14 05:33高浩华
石油与天然气化工 2016年5期
高浩华
神华集团北京低碳清洁能源研究所
渣油四组分吸附与裂化反应性能研究①
高浩华
神华集团北京低碳清洁能源研究所
利用四组分分离装置,在富集一定量四组分的基础上,通过测量四组分在催化剂上饱和吸附量的方法定量表征了四组分的吸附性能。此外,利用小型固定流化床实验装置,在常规反应条件下(反应温度500 ℃,剂油质量比6,重时空速20 h-1),考察了四组分的裂化反应性能。实验结果表明,四组分的吸附性能与裂化反应性能之间的排序存在较大的差异。饱和分裂化反应性能最强,其次为芳香分,胶质和芳香分裂化反应性能较为接近,沥青质最差;相比较裂化反应性能,胶质吸附性能最强,其次是芳香分,饱和分吸附性能最弱。
渣油四组分吸附性能裂化反应性能
催化裂化是典型的多相反应,各种烃类分子在催化剂表面的吸附是催化裂化化学反应过程的第1步,对整个催化裂化反应过程具有重要的影响[1-2]。由于不同种类的烃类分子在化学结构、分子尺寸和吸附热等方面的差异性[1-5],导致它们的吸附能力存在很大区别。根据现有研究表明[5-6],不同的烃类分子在催化剂表面吸附能力的强弱大致如下:稠环芳烃>稠环环烷烃>烯烃>单烷基侧链的单环芳烃>环烷烃>烷烃,且在同族烃类分子中,分子质量越大,则分子的吸附能力越强。但是,各种烃类化学反应能力大小顺序却与其吸附能力不相符合,大致情况为:烯烃>大分子单烷基侧链的单环芳烃>环烷烃>小单烷基侧链的单环芳烃>正构烷烃>稠环芳烃。因此,在催化裂化反应过程中,原料中吸附能力强而化学反应能力弱的烃类会优先占据有限的催化剂表面,由于它们难裂化且不易脱附,极大地阻碍了其他易于反应的烃类在催化剂表面的吸附和反应,从而存在明显的竞争吸附效应。
以上分析均基于单体烃类而言,鲜有对于渣油四组分反应和吸附性能直接研究报道。因此,本研究将利用自制的四组分分离装置,在富集一定量四组分的基础上,对四组分吸附和裂化反应性能进行了研究,相比较单组分烃类的竞争吸附而言,更加接近渣油的真实状况。
1 实验部分
1.1实验原料与催化剂
本研究以长庆减压渣油为研究对象,性质见表1。选用的催化剂为长庆重催装置工业平衡剂,主要物理化学性质列于表2。
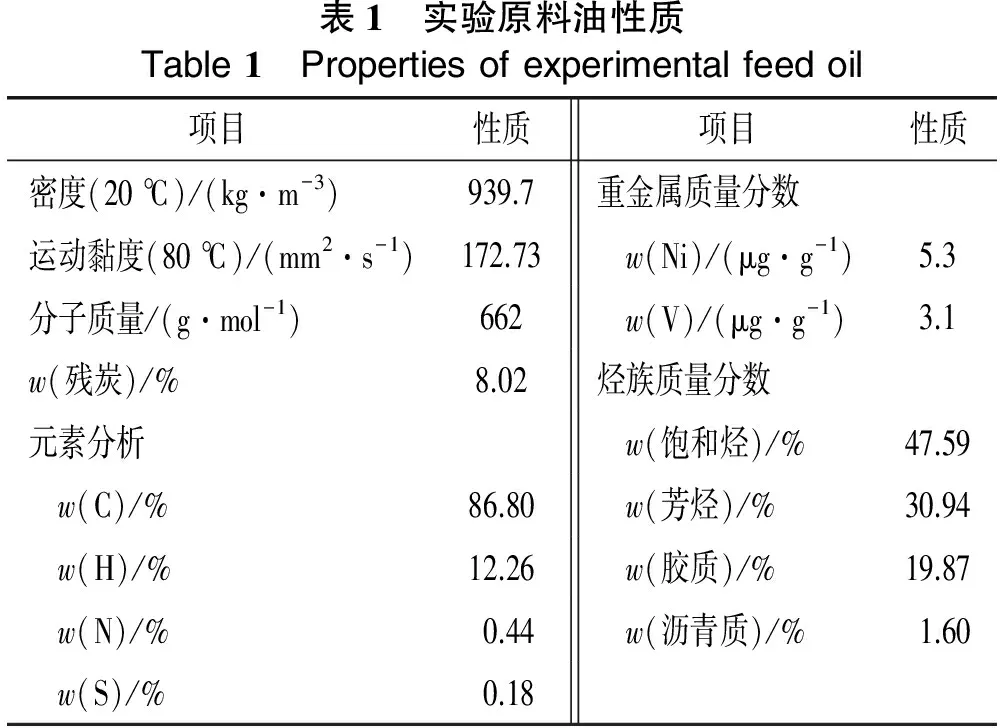
表1 实验原料油性质Table1 Propertiesofexperimentalfeedoil项目性质项目性质密度(20℃)/(kg·m-3)939.7重金属质量分数运动黏度(80℃)/(mm2·s-1)172.73 w(Ni)/(μg·g-1)5.3分子质量/(g·mol-1)662 w(V)/(μg·g-1)3.1w(残炭)/%8.02烃族质量分数元素分析 w(饱和烃)/%47.59 w(C)/%86.80 w(芳烃)/%30.94 w(H)/%12.26 w(胶质)/%19.87 w(N)/%0.44 w(沥青质)/%1.60 w(S)/%0.18

表2 LVR-60R催化剂性质Table2 PropertiesofcatalystsLVR-60R项目性质项目性质微反活性75 w(Na)/(μg·g-1)2487孔体积/(cm3·g-1)0.19 w(Fe)/(μg·g-1)3842比表面积/(m2·g-1)141粒径分布,w/%堆密度/(g·cm-3)0.98 0~40μm12.0金属质量分数 40~80μm45.8 w(Ni)/(μg·g-1)2155 >80μm42.2 w(V)/(μg·g-1)1419
1.2实验装置
为了能够获取大量四组分,将传统的四组分分离装置按比例进行放大,自制了一套分离四组分的实验装置,实验装置如图1所示。

实验在小型固定流化床实验装置上进行,关于装置及详细流程见文献[9]。采用Agilent 6890气相色谱仪测定实验收集的气体样品组成;采用Agilent 6890模拟蒸馏色谱仪分析液体产物,按其馏程定义C5~204 ℃馏分为汽油,204~350 ℃馏分为柴油,大于350 ℃馏分为重油;采用HIR-944B型红外碳硫分析仪测定待生催化剂上的焦炭含量。其中原料转化率按式(1)计算。
x=100%-重油产率
(1)
式中:x为原料转化率,%。
2 结果与讨论
2.1四组分吸附性能
由于催化裂化反应过程时间短,且反应温度高,几乎很难原位测量和表征催化裂化反应过程中的吸附现象。因此,本研究借鉴测定渣油饱和吸附量的方法[10],用于定量表征四组分的吸附能力的大小。为了在同一条件下比较单组分烃类的饱和吸附量,通常选用正庚烷或甲苯作为溶剂。由于沥青质不溶于正庚烷,仅溶于甲苯等芳烃类的溶剂中,如选用甲苯作为溶剂,因含有芳环,会严重影响饱和烃吸附量的测定。此外,对二者在重油中的含量进行比较,沥青质在重油中的含量远远低于饱和烃,权衡比较,本实验选用正庚烷作为溶剂进行饱和吸附量的测定,不对沥青质在催化剂的吸附情况进行表征。
实验方法及条件如下:
(1) 实验选用经过筛分40~80 μm的LVR-60R工业平衡剂为吸附催化剂,在700 ℃下,马弗炉焙烧2~3 h,放入干燥器冷却、干燥,备用。
(2) 实验选用正庚烷溶剂,将饱和分、芳香分、胶质配制成一定浓度的正庚烷溶液。
(3) 实验温度为20 ℃,加入10 g催化剂,在振荡器上以190次/min振荡24 h,由预先进行的探索性实验可知,此时已充分达到吸附平衡;将催化剂从溶液中取出,干燥,恒重,并对整个过程进行物料平衡的计算,吸附前后物料平衡大于95%(w),实验数据可用。为了减小实验误差,进行平行实验,求取平均值,实验结果如表3所示。
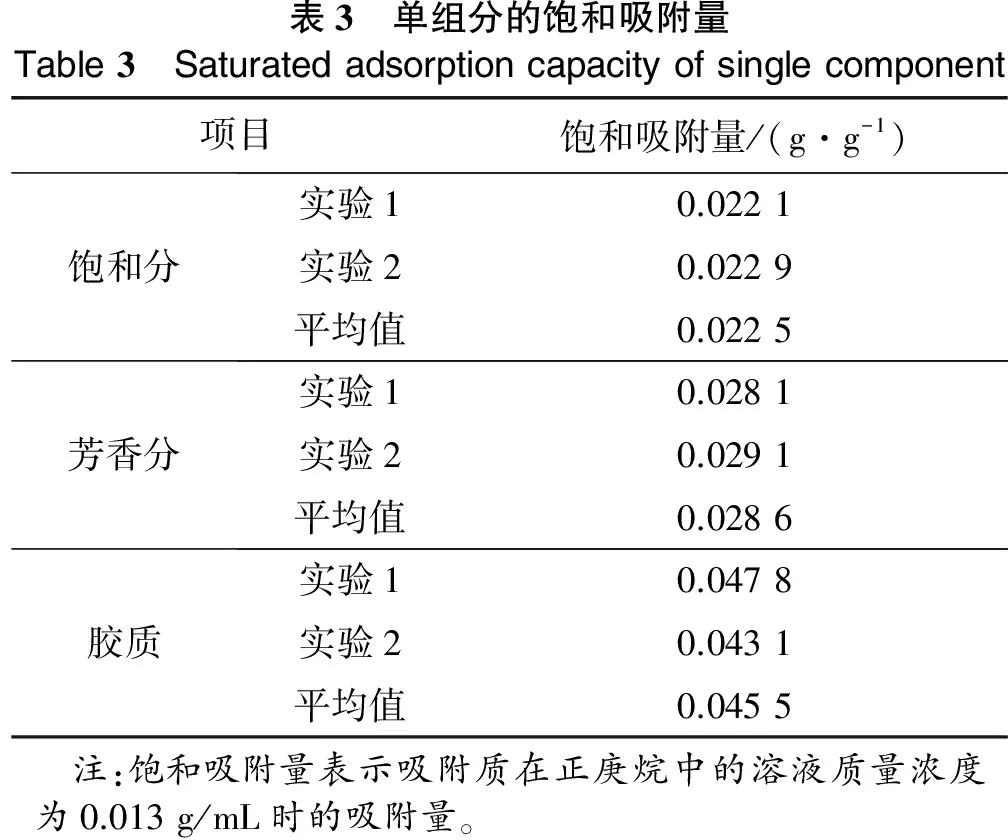
表3 单组分的饱和吸附量Table3 Saturatedadsorptioncapacityofsinglecomponent项目饱和吸附量/(g·g-1)饱和分实验10.0221实验20.0229平均值0.0225芳香分实验10.0281实验20.0291平均值0.0286胶质实验10.0478实验20.0431平均值0.0455 注:饱和吸附量表示吸附质在正庚烷中的溶液质量浓度为0.013g/mL时的吸附量。
本研究中吸附质在正庚烷中的溶液质量浓度为0.013 g/mL。根据溶剂稀释比对分离减压渣油沥青质的影响研究表明[6],当溶剂对油样的稀释比为40 mL/g时,溶液中不溶物沥青质的量达到稳定状态,且随着稀释比的增加不再发生变化。由此推测,当溶剂与胶质、芳香分的稀释比高于此比例时,胶质等能够稳定、充分地分散到正庚烷溶剂中。从表3可以看出,不同族组成的烃类饱和吸附量存在较大的区别。在溶液质量浓度为0.013 g/mL时,胶质的饱和吸附量最高,为0.045 5 g/g,其次是芳香分,为0.028 6 g/g,最后是饱和分,为 0.022 5 g/g。饱和吸附量的差异可以从其组成结构得到进一步的解释。
表4列出我国几种典型的减压渣油四组分性质[6]。从表4可以看出,饱和分分子质量一般在500~900 g/mol之间,基本由烷烃和环烷烃组成,且环烷烃的环数一般也集中于1~3环之间;芳香分的分子质量一般略高于饱和分,约在600~1 100 g/mol之间,基本上以1~3环的芳香烃为主,并含有部分非烃类化合物[6],这些芳香烃的结构中含有相当多的烷基碳,且烷基碳的数目也通常大于芳香碳的数目,同时还往往并合或连有环烷环;胶质的平均分子质量一般在1 000~2 800 g/mol之间,是石油中组成结构最复杂的组分,平均每个分子结构中含有2个以上的杂原子[6],芳香环数一般大于5,总环数在7~10之间,实验结果说明,随着分子质量、分子极性和芳香度的增加,其饱和吸附量逐渐增加,实验结果很好地验证了前面的理论分析。无论是从单体烃,还是四组分的吸附性能方面,都充分证实了四组分吸附性能的差异性。
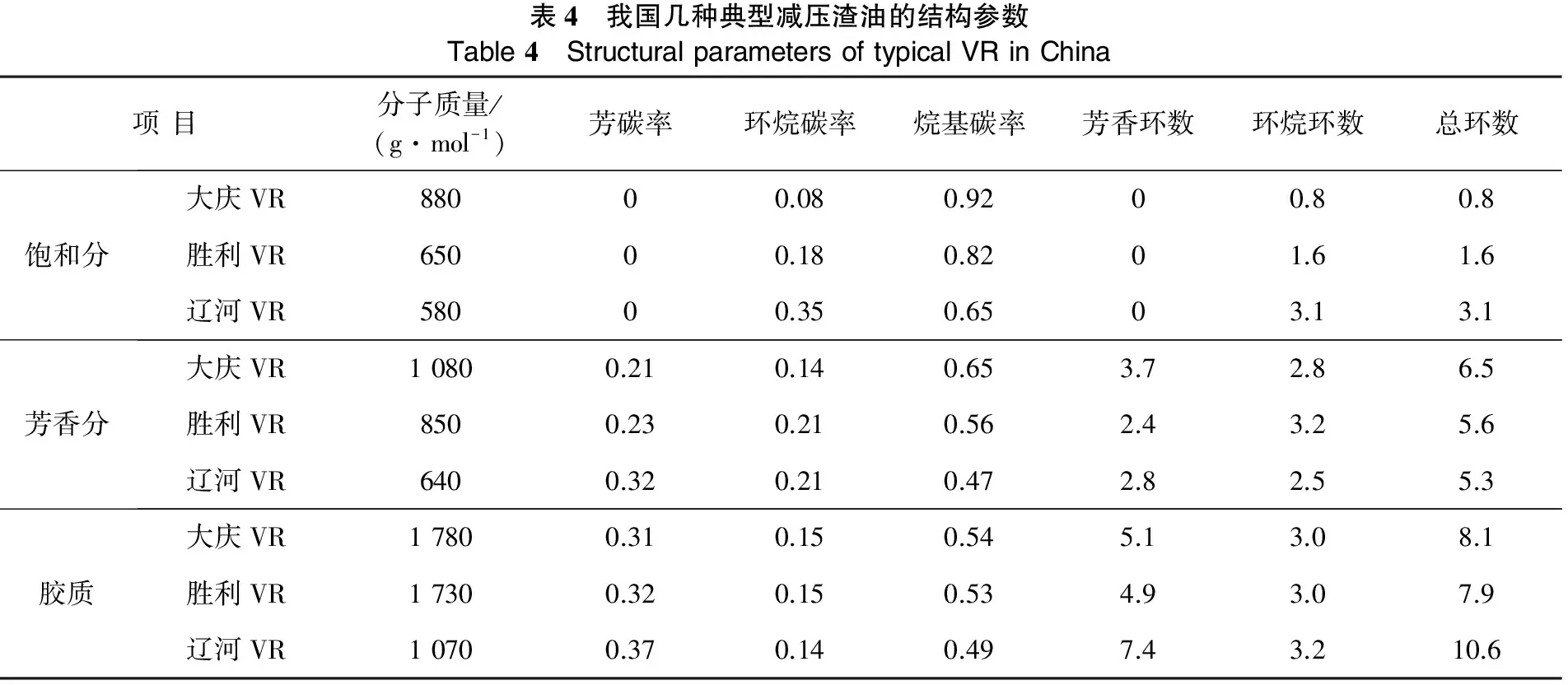
表4 我国几种典型减压渣油的结构参数Table4 StructuralparametersoftypicalVRinChina项目分子质量/(g·mol-1)芳碳率环烷碳率烷基碳率芳香环数环烷环数总环数饱和分大庆VR88000.080.9200.80.8胜利VR65000.180.8201.61.6辽河VR58000.350.6503.13.1芳香分大庆VR10800.210.140.653.72.86.5胜利VR8500.230.210.562.43.25.6辽河VR6400.320.210.472.82.55.3胶质大庆VR17800.310.150.545.13.08.1胜利VR17300.320.150.534.93.07.9辽河VR10700.370.140.497.43.210.6
2.2四组分的催化裂化反应性能
由于芳香分和胶质流动性差,沥青质呈固态的特性,因此,为了改善原料的流动性,本实验将甲苯搀兑到芳香分、胶质和沥青质中,进行催化裂化反应性能的评价。根据催化裂化反应的正碳离子反应机理发现,生成甲基正碳离子需要很高能量。因此,在催化裂化条件下,甲苯的裂解比较困难,一般情况下不会发生开环反应,而主要发生歧化反应[11]。从理论上来说,利用甲苯作为搀兑溶剂用于四组分反应性能的评价是可行的,且甲苯与芳香分、胶质和沥青质相比极性也很小,不会对芳香分、胶质和沥青质中的芳香烃类产生竞争吸附作用。为了验证以上的分析,在常规的反应条件下(反应温度500 ℃,剂油质量比(以下简称剂油比)为6,重时空速20 h-1),采用LVR-60R平衡剂作为催化剂,考察了甲苯单独反应时的产品分布,如表5所示。
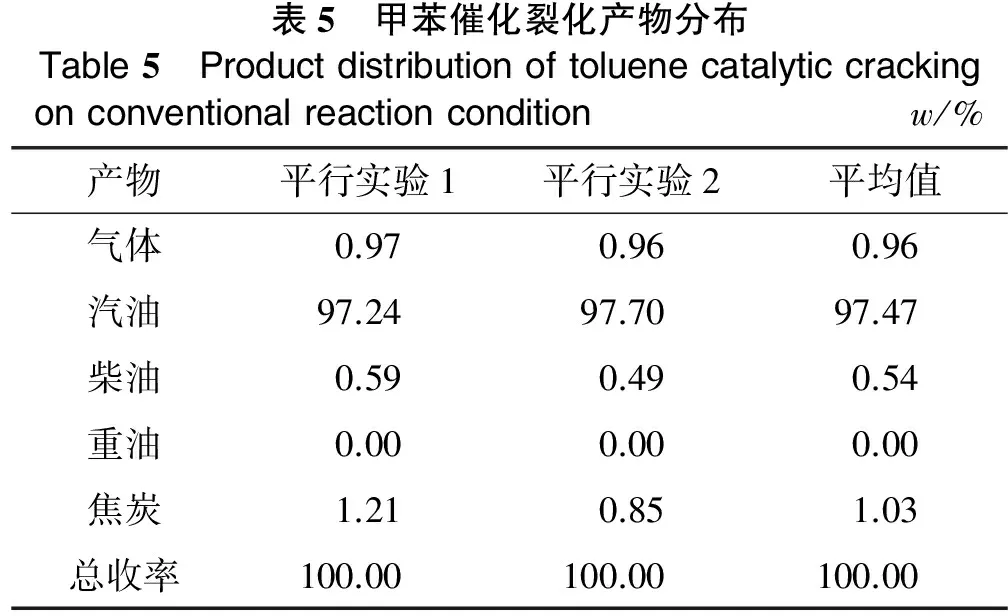
表5 甲苯催化裂化产物分布Table5 Productdistributionoftoluenecatalyticcrackingonconventionalreactionconditionw/%产物平行实验1平行实验2平均值气体0.970.960.96汽油97.2497.7097.47柴油0.590.490.54重油0.000.000.00焦炭1.210.851.03总收率100.00100.00100.00
从表5可以看出,甲苯催化裂化液体产物产率达到98%(w)以上,气体产物不到1%(w),焦炭产率为1.03%(w)。气体色谱分析表明,气体中大部分为空气,还有极少量的丙烯、丁烯和氢气。对液体产物进行质谱分析,发现除甲苯外,液体产物中有多种芳烃的存在,主要为苯和二甲苯,还有极少量两个苯环的烃类,故将甲苯作为溶剂搀兑到芳香分、胶质和沥青质中用于催化裂化反应性能的评价是可行的。
在获取大量四组分的基础上,利用小型固定流化床实验装置,在常规反应条件下,LVR-60R工业平衡剂作为催化剂,考察了饱和分、芳香分+甲苯(质量比为1∶2)、胶质+甲苯(质量比1∶2)和沥青质+甲苯(质量比1∶4)的催化裂化反应性能。为了减小实验误差,对以上实验进行了平行实验,芳香分、胶质和沥青质单独反应时的产物分布则通过差减法计算得出,计算结果见表6。
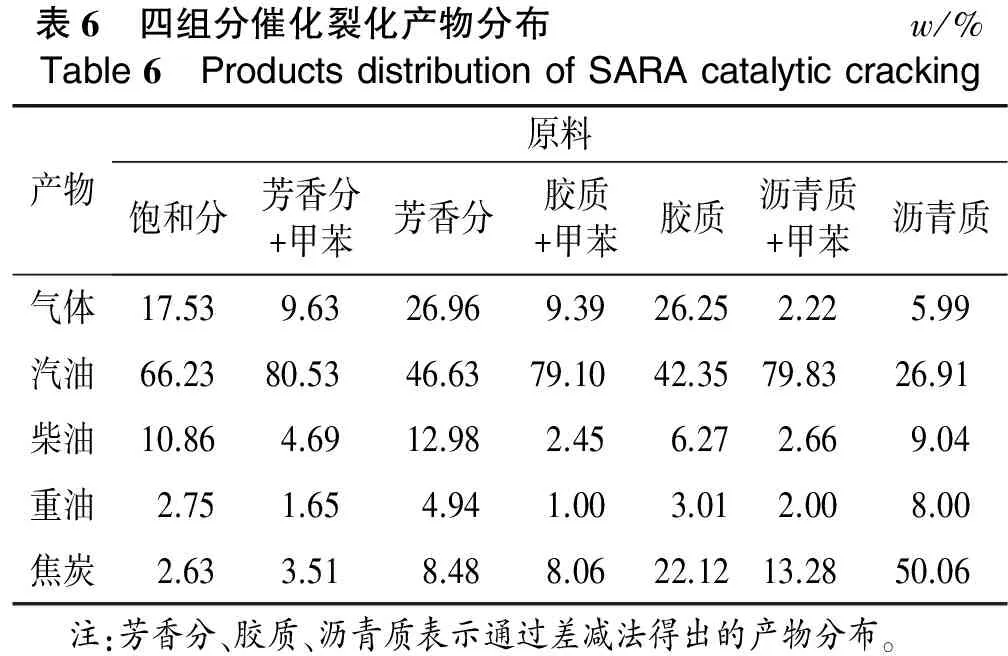
表6 四组分催化裂化产物分布w/%Table6 ProductsdistributionofSARAcatalyticcracking产物原料饱和分芳香分+甲苯芳香分胶质+甲苯胶质沥青质+甲苯沥青质气体17.539.6326.969.3926.252.225.99汽油66.2380.5346.6379.1042.3579.8326.91柴油10.864.6912.982.456.272.669.04重油2.751.654.941.003.012.008.00焦炭2.633.518.488.0622.1213.2850.06 注:芳香分、胶质、沥青质表示通过差减法得出的产物分布。
从表6可以看出,四组分裂化的产物分布存在巨大的差异,但均具有很高的转化率。表明饱和分本身极易裂化,在没有多环芳烃和极性非烃类对竞争吸附作用下,饱和分表现出优良的反应性能;对于芳香分、胶质和沥青质,通过搀兑甲苯进料,极大地改善了芳香分、胶质和沥青质的流动性,有利于与催化剂的接触,从而加大了原料的裂化深度,并获得较高的转化率。
从产物分布来看,实验结果与在微型反应-色谱联合实验装置上获得的实验数据相接近[12]。饱和分轻质油收率最高,达到77%(w)以上,其次是芳香分、胶质,最后是沥青质;饱和分的焦炭收率最低,仅为2.63%(w),而有超过50%(w)沥青质脱氢、缩合生成焦炭;芳香分和胶质裂化气体产物较高,达到26%(w)以上,饱和分为17.53%(w),沥青质仅为5.99%(w)。四组分裂化产物中,柴油产物均较少,但芳香分的柴油收率最高,为12.98%(w),这是由于芳环在裂化反应时一般不开环。因此,芳香分在裂化时一般只发生断侧链、或与芳环相连的环烷环发生开环裂化反应,而剩余的芳环则作为一个整体由于碳数的降低而落入柴油馏分,使得芳香分裂化时柴油馏分的收率明显高于其他组分。
从芳香分和胶质的裂化产物分布可以看出,芳香分和胶质均具有较好的产物分布,它们的气体、汽油收率较为接近;胶质的轻质油收率也达到48%(w)以上,表明胶质并不像人们想象的那样大部分将脱氢、缩合生成焦炭,依然具有较好的可裂化性能。芳香分和胶质可看成3部分组合,芳香环部分、环烷环部分和烷基侧链部分,而二者的最大区别在于芳香环和环烷环的环数。研究表明,芳香分的环数绝大部分小于3个,而胶质的平均芳环数已接近5个[6],因此,如前所述,对于芳香分,这些芳环经断侧链和环烷环后极易落入轻质油馏分,而胶质中的芳香环在不发生开环的情况下已经很难形成气相产物被带出,最终只能缩合成焦炭,这也是胶质的焦炭收率明显高于芳香分的重要原因。
通过对四组分裂化反应性能的评价,证实它们的反应性能之间存在很大的差异;饱和分具有极高的可裂化性能,经裂化绝大部分生成轻质油和气体;芳香分和胶质的产物相对比较接近。相比芳香分,胶质的焦炭收率明显增加,但胶质也并不像人们想象的那样大部分缩合生成焦炭,焦炭收率仅为22.12%(w);沥青质绝大部分生产焦炭,虽然在重油中的含量较低,但其稠环芳烃和重金属组成的复杂结构对饱和分的裂化会导致严重的阻滞作用。
此外,通过比较四组分反应、吸附能力的大小还发现,吸附能力强的胶质烃类,反应性能较差,由于吸附能力强,经裂化生产的产物不易脱附,导致产物中终端产物含量较高;而饱和分则与之完全相反,由于吸附能力较差,说明在催化裂化反应过程中,烃类分子经反应生产小分子烃类后,容易从活性中心脱附,减少进一步生焦和二次裂化的可能性,其产物以轻质油为主,这与前面对单体烃的反应和吸附能力大小的分析相吻合。
3 结 论
实验研究证实了四组分吸附性能与裂化反应性能之间存在较大的差异。饱和分具有极好的裂化反应性能,轻质油和气体收率超过90%(w),焦炭和重油收率小于10%(w);芳香分和胶质的产物分布比较接近,轻质油和气体产物的收率在74%~87%(w)之间,但胶质的焦炭收率则远大于芳香分;沥青质的裂化反应性能最差,焦炭收率大于50%(w)。相比四组分的裂化反应性能,四组分的吸附性能与之相反,胶质具有最强的吸附能力,其次是芳香分,饱和分吸附能力最弱。
[1] PRUSKI J, PEKEDIZ A, DE LASA H. Catalytic cracking of hydrocarbons in a novel riser simulation: lump adsorption parameters under reaction conditions[J]. Chemical Engineering Science, 1996, 51(10): 1799-1806.
[2] CORMA A, ORTEGA F J. Influence of adsorption parameters on catalytic cracking and catalyst decay[J]. Journal of Catalyst, 2005, 233(2): 257-265.
[3] LI B, CALEMMA V, GAMBARO C, et al. Competitive adsorption of C20-C36 linear paraffins on the amorphous microporous silica-alumina ERS-8 in vapor phase and liquid phase[J]. Industrial & Engineering Chemistry Research, 2010, 49(16): 7541-7549.
[4] MARTIGNONI W, DE LASA H. Heterogeneous reaction model for FCC riser units[J]. Chemical Engineering Science, 2001, 56(2): 605-612.
[5] GATES B C, KATZER J R, SEHUIT G C A. Chemistry of Catalytic Processes[M]. New York: McGraw-Hill Book Company, 1979.
[6] 梁文杰. 重质油化学[M]. 东营: 石油大学出版社, 2000.
[7] REYES S C. Analysis of zeolite catalyst deactivation during catalytic cracking reactions[J]. Industrial & Engineering Chemistry Research, 1991, 30(1): 71-82.
[8] LI Z K, GAO J S, WANG G, et al. Influence of nonbasic nitrogen compounds and condensed aromatics on coker gas oil catalytic cracking and their characterization[J]. Industrial & Engineering Chemistry Research, 2011, 50(15): 9415-9424.
[9] GAO H H, WANG G, LI R, et al. Study on the catalytic cracking of heavy oil by proper cut for higher conversion and desirable products[J]. Energy & Fuels, 2012, 26(3): 1880-1891.
[10] LIU Z Y, CHEN S L, GE X J, et al. Measurement of Diffusion Coefficient of Heavy Oil in Fluidized Catalytic Cracking (FCC) Catalysts[J]. Energy & Fuels, 2010, 24(5): 2825-2829.
[11] DE LA PUENTE G, DEVARD A, SEDRAN U. Conversion of residual feedstocks in FCC. evaluation of feedstock reactivity and product distributions in the laboratory[J]. Energy & Fuels, 2007, 21(6): 3090-3094.
[12] 徐春明, 高金森, 林世雄, 等. 重油催化裂化反应过程分析[M]. 北京: 石油工业出版社, 2002.
Study on the adsorbability and crackability of resid SARA
Gao Haohua
(NationalInstituteofClean-and-Low-CarbonEnergy,ShenhuaGroupCorporationLimited,Beijing102211,China)
By using self-made SARA separation device, amounts of SARA were obtained. The adsorbability of SARA was determined by measuring the saturated adsorption amount on the catalyst. The crackability of SARA was carried out in the fixed fulided reactor under conventional reaction conditions (reaction temperature of 500 ℃, catalyst-to-oil weight ratio (CTO) of 6, weight hourly space velocity (WHSV) of 20 h-1). The results confirmed that the huge differences are existed between the order of adsorbability and crackability of resid SARA. The crackability of saturate is the best, followed by aromatic; the crackability of resin is close to that of aromatic; the worst is asphaltene. Compared to the crackability of SARA, the resin has the strongest adsorbability, followed by aromatic; the saturate is the weakest.
resid, SARA, adsorbability, crackability
高浩华(1983-),男,湖南常德人,博士,现就职于神华集团北京低碳清洁能源研究所,主要从事油品加工工艺及催化剂开发。E-mail:gaohaohua_530@163.com
TE624.9
ADOI: 10.3969/j.issn.1007-3426.2016.05.002
2016-03-31;编辑:温冬云
猜你喜欢
中华养生保健(2020年9期)2021-01-18
少儿美术(2019年8期)2019-12-14
无机化学学报(2019年2期)2019-02-27
石油石化绿色低碳(2019年6期)2019-01-14
石油石化绿色低碳(2019年6期)2019-01-14
少儿美术(快乐历史地理)(2018年7期)2018-04-02
岭南音乐(2017年3期)2017-07-18
当代化工研究(2016年6期)2016-03-20
化工进展(2015年6期)2015-11-13
传奇故事(破茧成蝶)(2015年1期)2015-02-28
