
基于高通量测序分析青藏高原特有植物蓝玉簪龙胆(Gentiana veitchiorum)的SSR和SNP特征
2016-11-10 10:49田尊哲高庆波陈世龙张发起
植物研究 2016年5期
田尊哲 高庆波 陈世龙 张发起*
(1.中国科学院高原生物适应与进化重点实验室,中国科学院西北高原生物研究所,西宁 810001; 2.中国科学院大学,北京 100039)
基于高通量测序分析青藏高原特有植物蓝玉簪龙胆(Gentianaveitchiorum)的SSR和SNP特征
田尊哲1,2高庆波1陈世龙1张发起1*
(1.中国科学院高原生物适应与进化重点实验室,中国科学院西北高原生物研究所,西宁 810001;2.中国科学院大学,北京 100039)
利用Illumina HiSeqTM2500平台对青海省库泽县的蓝玉簪龙胆(Gentianaveitchiorum)进行高通量测序,共得到SSR序列8 588条,对其SSR重复类型进行分析;三核苷酸重复类型所占的比例最大占54.7%(4696);其次是二核苷酸重复类型占41.3%(3543);四核苷酸重复类型,五核苷酸重复类型和六核苷酸重复类型所占的比例较少(共占4%)。在二核苷酸重复类型中AT/TA重复类型所占的比例最大分别为9.85%和9.5%。在微卫星中重复单元的长度大小和重复次数成负相关,并且微卫星的总长度与重复单元的长度成正相关。在蓝玉簪龙胆的花(GP-F)和叶(GP-L)中分别得到253 789和249 417个SNP位点,其中在非编码区上的比例为51.29%和51.96%。分析发现蓝玉簪龙胆SNP位点在编码区中同义转换所占的比例(48.63%和47.96%)要远远高于非同义转换的比例(0.08%和0.08%),可能与功能基因序列相对稳定有关。
蓝玉簪龙胆;高通量测序;微卫星;单核苷酸多态性;青藏高原
青藏高原(Qinghai-Tibetan Plateau)是世界上海拔最高的高原,平均海拔达到4 500 m,面积达到2.5×106km2,被誉为“世界屋脊”、“地球的第三极”[1]。蓝玉簪龙胆(GentianaveitchiorumHemsl.)为龙胆科(Gentianaceae)龙胆属(Gentiana)的多年生草本植物,在青藏高原地区广泛分布,高5~10 cm,营养叶和繁殖叶异型。龙胆花主要含有裂环环烯醚类活性成分,如:落甘酸、獐芽菜苦苷、龙胆苦苷和獐牙菜苷等,其叶具有清湿热、泻肝胆湿火、镇咳利喉健胃的功能,主治感冒发烧目赤咽痛肺热咳嗽等[2]。对蓝玉簪龙胆的研究主要集中在化学成分以及药效等方面[3~5],而在分子遗传等方面研究较少。青藏高原的温度从1960年就开始升高,这比中国其他地方的地区都要早[7~8],并且在过去的50年中,青藏高原每十年温度就要增高0.3℃,比中国其他地区的温度都要上升的快[5]。青藏高原的环境变化对生物多样性有了很大的影响,为了更加深入的挖掘蓝玉簪龙胆资源和建立完善的种质资源评估和保护系统,因此很有必要开展蓝玉簪龙胆的遗传多样性研究。
近年来,随着分子技术的快速发展,尤其是第二代微卫星标记和第三代单核苷酸标记在探究生物群体内和群体间遗传变异及种间关系和遗传育种等研究中所凸显的优越性,使的其运用范围越来越广。微卫星,又称简单重复序列标记(simple sequence repeats,SSRs),是一类由几个核苷酸(2~6个)为重复单位组成的长达几十个核苷酸的重复序列,长度较短,且广泛分布于真核生物的基因组中,具有数量多,分布广且均匀,高度多样性,分析快速方便等优点[9~12]。此外,微卫星序列在群体中通常具有很高的多态性,而且一般为共显性,这些特点使得微卫星标记成为分子遗传研究中使用最为广泛的遗传标记之一。
单核苷酸多态性(Single Nucleotide Polymorphism,SNP)是指一物种不同个体在基因组水平上有单个核苷酸变异引起的DNA序列多态性,通常被认为是变异在频率大于1%时称为SNP[13]。SNP一般只涉及到碱基的转换(Transition)和颠换(Transversion),是许多真核生物中最丰富的遗传变异方式,具有数量多,分布广,突变率低,易实现自动化检测等优点[14]。
本研究以选取采集于青海泽库蓝玉簪龙胆的叶和花开展高通量测序,挖掘蓝玉簪龙胆的SSR和SNP信息,并分析其在该物种的特征,为蓝玉簪龙胆的遗传多样性以及群体遗传学的研究奠定了分子基础。
1 样品的采集和方法
1.1 样品的采集和高通量测序、拼接
蓝玉簪龙胆(G.veitchiorumHemsl;Zhang 2014131)采集于青海省泽库县(35°4′N,101°30′E,3 681 m)。采取同一株上的叶片和花后放入液氮中保存,带回实验室于-80℃保存。凭证标本保存于中国科学院西北高原生物研究所青藏高原生物标本馆(HNWP)。
从蓝玉簪龙胆的叶(GP-L)和花(GP-F)中各提取100 μg总RNA;用Nanodrop检测RNA的纯度,用Qubit对RNA浓度进行精确定量,并用Agilent 2100精确检测RNA的完整性;然后用Oligo(dT)的磁珠富集蓝玉簪龙胆的mRNA;随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs和DNA polymeraseⅠ和RNase H合成双链cDNA,再用AMPure XP beads纯化双链cDNA。纯化的双链cDNA先进行末端修复,加A尾并连接测序接头,再用AMPure XP beads进行片段大小选择。最后进行PCR扩增,并用AMPure XP beads纯化PCR产物,得到最终的文库。文库合格后,把不同文库按照有效浓度及目标下机数据量的需求混池后在Illumina HiSeqTM2500平台上测序。
对所得的数据进行质量评估,包括:测序错误率分布检查,A/T/G/C的含量分布检查,其中对单个碱基位置的错误率控制在1%以下,然后对测得的原始测序序列(raw reads)进行过滤:去除带接头(adapter)的和N的比例大于10%的reads,并去除低质量reads,得到干净的读序(clean reads)后,而后用Trinity[15]对得到的clean reads进行拼接以获取后续分析的参考序列,并取每条基因中最长的转录本作为Unigene,以此进行后续分析。
1.2 SSR和SNP的检测,筛选和统计分析
基于蓝玉簪龙胆转录组的SSR检测是以组装出来的Unigene作为参考序列,采用MicroSatellite(MISA;http://pgra.ipk-gatersleben.de/misa/)对Unigene进行SSR检测。SSR搜索标准有精确型(perfect)及复合型(compound)的SSR重复单元[16],各重复微卫星类型重复次数设定为:两碱基(dinnucleotide repeats,DNRs)至少重复6次,三碱基(trinucleotide repeats,TNRs)至少重复5次,四碱基(tetranucleotide repeats,TTNRs)至少重复5次,五碱基(pentanucleotide repeats,PTNRs)至少重复4次,六碱基(hexanucleotide repeats)至少重复4次。并对蓝玉簪龙胆转录本的不同SSR类型以及SSR类型中密度进行统计分析。
通过samtools和piacard-tools等工具对比对结果进行染色体坐标排序,去掉重复的reads等处理,最后通过变异检测软件GATK2[17]以Unigene为参考序列对resds进行SNP Calling,并对原始结果进行过滤(过滤掉质量值小于30,距离小于5的SNP)。并对得到的SNP位点位于编码区或者非编码区,以及在编码区中的同义转换或者非同义转换的SNP数量进行统计。
2 结果与分析
2.1 测序产量,质控和组装结果
随着高通量测序技术的发展及成本的降低,通过高通量测序可以得到海量的序列,但是在RNA-seq技术中,测序错误率会随着测序序列长度的增加而升高[18~19],在单个碱基位置的测序错误率控制在1%以下,在GP-F和GP-L的单碱基错误率都为0.03%;在由于测序得到的reads并不都是有效的,里面含有带接头的,重复的和测序质量低的reads,它们对组装及后续分析都会产生影响。在对GP-F和GP-L的转录本测得的raw reads去除含接头的、N的比例大于10%的以及低质量的reads后。分别得到22 936 570条和22 355 444条clean reads,分别占Raw reads的92.80%和92.59%,在两个GP-F和GP-L中质量值≧20%的碱基数占总碱基数的比例分别为95.72%和95.96%,质量值≧30%的碱基数占总碱基数的比例分别为91.81%和92.14%,碱基G和C含量总和分别为43.23%和42.79%以上。
在对GP-F和GP-L的raw reads进行处理后,我们用组装软件Trinity对所得的clean reads进行组装,对转录本和Unigene的长度进行统计(图1),其中在拼接得到的Transcripts中N50为1 424,而N90为314;在拼接得到的Unigene中N50为910,而N90为256。其中拼接的Transcripts和Unigene总的核苷酸数分别为177 300 983个和85 642 977个。

图1 拼接后的Unigene与Transcript长度分布图Fig.1 The length distribution of Unigene and Transcript after assembled
2.2SSR的测序结果,测序产量、质量及组装结果分析
在Illumina HiSeqTM2500测序平台对蓝玉簪龙胆测序后,去除Raw reads中含有接头的,N含量比大于10%的和低质量的reads后,用Trinity对clean reads进行拼接。在对Trinity拼接得到的转录序列为参考序列,取每条基因中最长的转录本作为Unigene。通过对Unigene进行SSR检测,共检测的序列共140 225条,总长度达到85 642 977 bp,共检测到8 588条SSR序。
通过对SSR类型进行分析发现:双核苷酸重复类型SSR占41.3%(3 543个),长度以12~24 bp为主;三核苷酸重复类型SSR占54.7%(4 696个),长度以15~24 bp为主;四核苷酸重复类型SSR占3.4%(296个),长度以20~24 bp为主;五核苷酸重复类型和六核苷酸重复类型SSR较少(图2)。对其进一步统计分析得到二核苷酸重复类型有12种,三核苷酸重复类型有60种,四核苷酸重复类型有92种,五核苷酸重复类型21种,六核苷酸重复类型有32种(图3)。

图2 蓝玉簪龙胆不同核苷酸重复类型的SSR数量Fig.2 Number diversification of SSR in nucleotide repeat sequence of G.veitchiorum
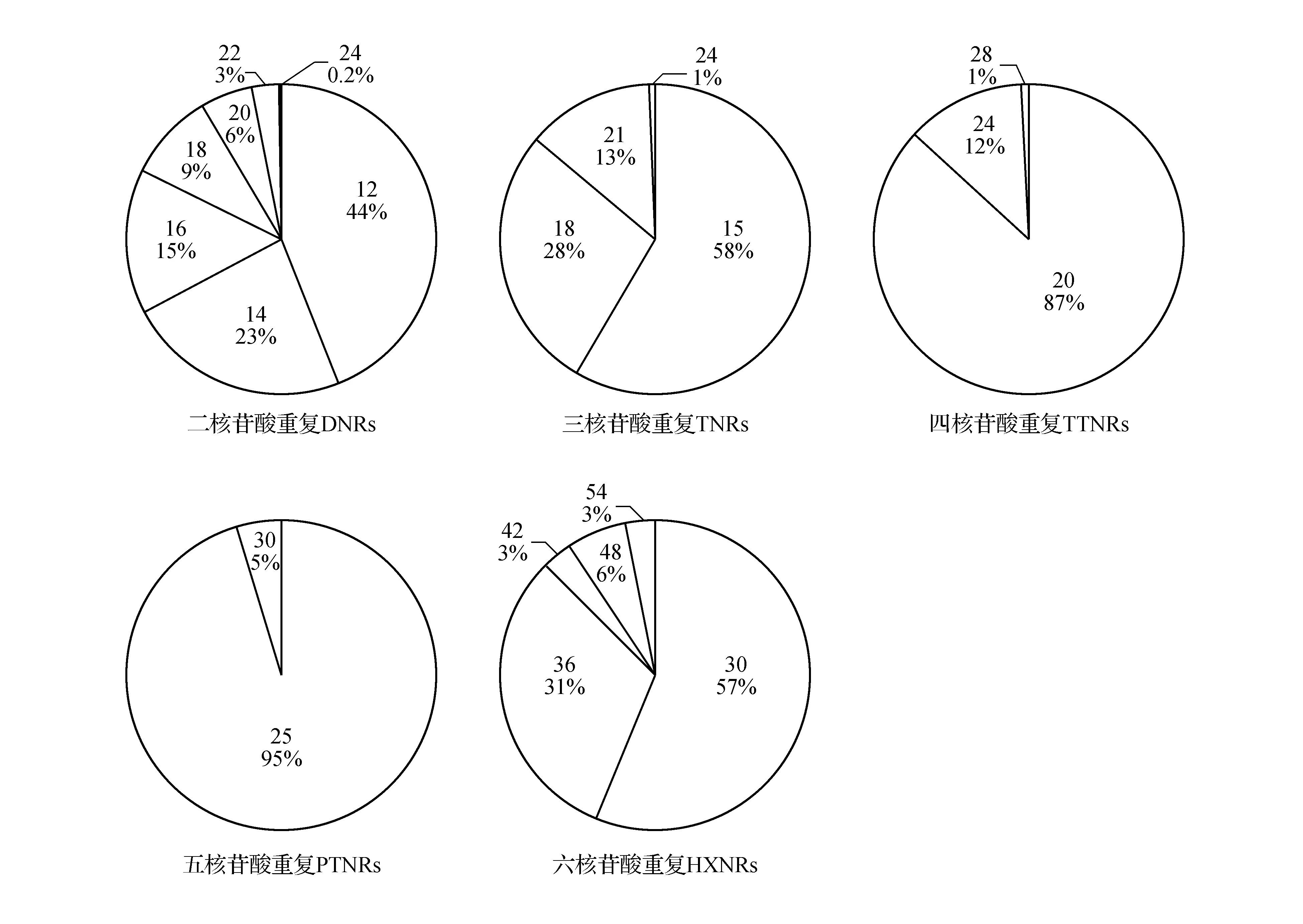
图3 蓝玉簪龙胆序列不同长度重复类型微卫星长度分布及其变异 每一扇形区对应不同长度微卫星所占百分比,上面的值为对应的微卫星片段长度。Fig.3 Diversification of the microsatellites in G.veitchiorum sequence of nucleotide repeat Each sector represents the percent of ditterent SSR,the number is the lengh of the SSR.
通过对主要的核苷酸重复类型的比较分析发现,在二核苷酸重复类型中,AT/TA重复类型所占比例较高,AC/TG次之;在三核苷酸重复类型中GAA重复类型所占比例较高,AGA/TCT重复类型所占比例次之;四核苷酸重复类型、五核苷酸重复类型、六核苷酸重复类型的数量相对较少(表1)。
2.3 蓝玉簪龙胆的SNP位点特征分析
通过samtools和picard-tools等工具比对GP-F和GP-L结果进行染色体坐标排序,去掉重复的reads等处理后,以Unigene序列为参考序列,通过变异检测软件GATK2进行SNP Calling,并对原始结果进行过滤,在GP-F和GP-L中分别得到253 789和249 417个SNP位点,其中非编码区的SNP位点分别为130 175个和129 593个,编码区的SNP位点分别为123 614个和119 824个。其中在CP-F中编码SNP中,同义转换占48.63%(123 431个)非同义转换占0.08%(201个);在GP-L中的编码区的SNP中,同义转换占47.96%(119 620),非同义转换占0.08%(201个;表2)。对其进一步分析,GP-F和GP-L的每个SNP的平均跨度分别为699和711 bp。在GP-F和GP-L中的SNP的位点个数和跨度出现此差别表明在蓝玉簪龙胆在适应环境变化过程中,花的变异量强于叶的变异量,或者可能是由于花所受环境变化的影响更大。
对蓝玉簪龙胆SNP类型分析发现,在GP-F中转换类型占58%(147 198个),颠换类型占42%(110 180个);在GP-L中转换类型占61%(52 144个),颠换类型占39%(97 272个,表3)。其中在GP-F和GP-L中的转换类型明显高于颠换类型,在GP-F和GP-L中的转换类型中,C<->T发生频率较高,这可能与SNPs在CG序列上出现的最为频繁,而C(胞嘧啶)常以甲基化形式存在,在脱氨后即成为T(胸腺嘧啶)有关[20],在蓝玉簪龙胆的花和叶中的SNPs类型及其发生频率趋势基本一致。
表1主要核苷酸重复序列类型在蓝玉簪龙胆序列的百分比
Table1RelativepercentageofSSRsinG.veitchiorumsequences
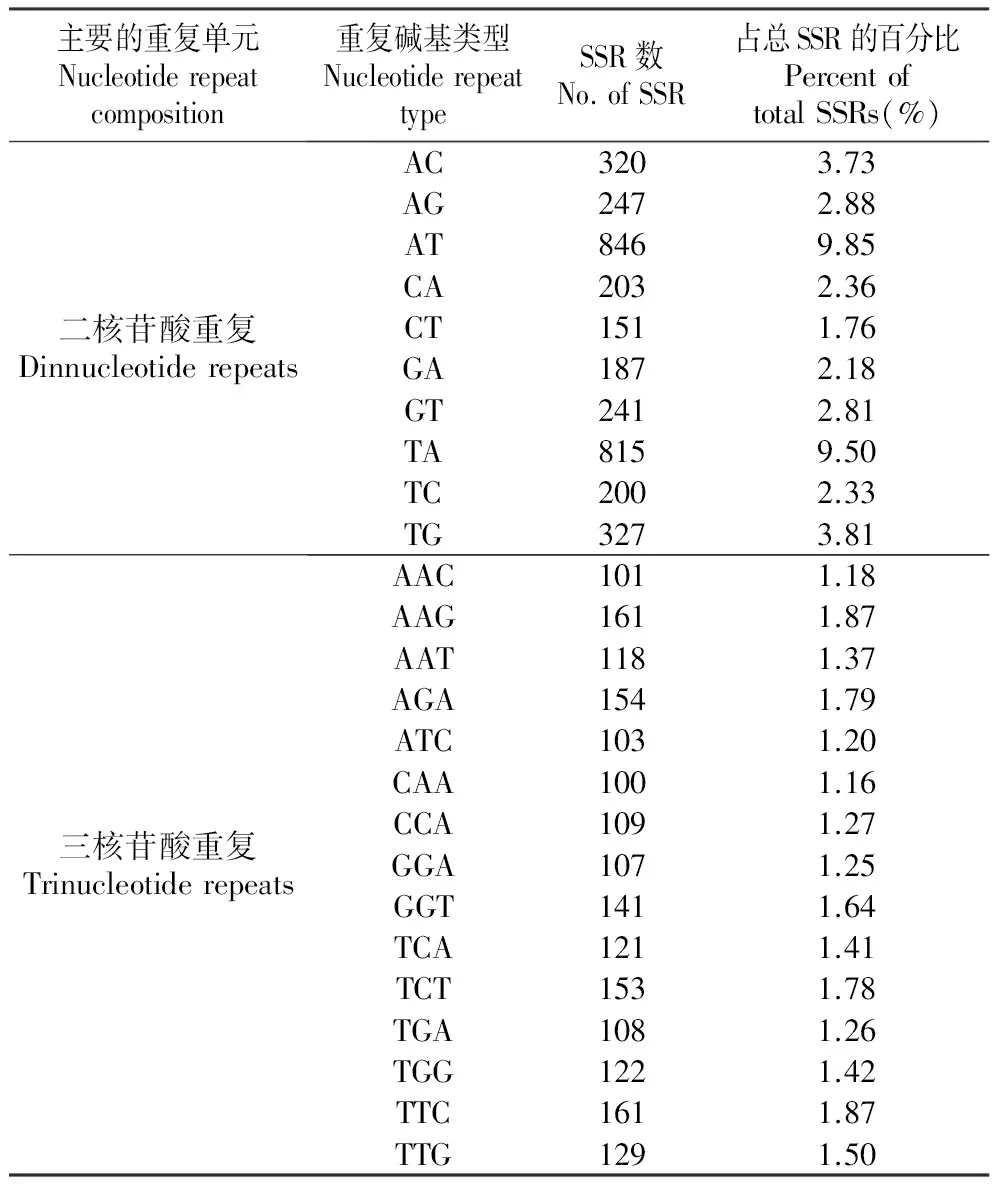
主要的重复单元Nucleotiderepeatcomposition重复碱基类型NucleotiderepeattypeSSR数No.ofSSR占总SSR的百分比PercentoftotalSSRs(%)二核苷酸重复DinnucleotiderepeatsAC3203.73AG2472.88AT8469.85CA2032.36CT1511.76GA1872.18GT2412.81TA8159.50TC2002.33TG3273.81三核苷酸重复TrinucleotiderepeatsAAC1011.18AAG1611.87AAT1181.37AGA1541.79ATC1031.20CAA1001.16CCA1091.27GGA1071.25GGT1411.64TCA1211.41TCT1531.78TGA1081.26TGG1221.42TTC1611.87TTG1291.50
表2高通量测序鉴定的蓝玉簪龙胆的SNPs类型分类
Table2IdentifiedtheSNPstypeofG.veitchiorumbyhigh-throughputsequencing

样品Sample总的SNP位点数Totalno.ofSNP非编码区SNPNo⁃codingSNP编码区SNPCodingSNP同义转换Synonymoustransition非同义转换NonsynonymoustransitionGP⁃F253789(100%)130175(51.29%)123614(48.71%)123413(48.63%)201(0.08%)GP⁃L249417(100%)129593(51.96%)119824(48.04%)119620(47.96%)201(0.08%)
表3高通量测序鉴定的蓝玉簪龙胆SNPs类型分析
Table3AnalysistheSNPstypeofG.veitchiorumbyhigh-throughputsequencing

样品SampleSNP类型SNPtype数量NumberSNP类型SNPtype数量NumberGP⁃F转换transition颠换transversionA<->G73590A<->T37596C<->T73608G<->T21064——C<->G14974——A<->C36546合计147198(58%)合计110180(42%)GP⁃LA<->G77319A<->T34918C<->T74825G<->T22448——C<->G16960——A<->C22946合计152144(61%)合计97272(39%)
3 讨论
通过Illumina HiSeqTM2500测序平台对蓝玉簪龙胆转录组测序共得到8 588条SSR序列,平均跨度为9 973 bp,在蓝玉簪龙胆基因组中的SSR类型中,三核苷酸重复类型所占的比例最大(54.7%),其次是二核苷酸重复类型(41.3%),而在同属于龙胆亚族的川西獐牙菜(Swertiamussotii)的SSR的平均跨度为12.6 kB,在SSR重复类型中三碱基重复所占的比例为45.99%,二碱基重复类型所占的比例为41.62%[21]。其中三碱基重复类型的比例相差比较大,此结果说明即使在同一亚族中不同属之间的SSR类型分布比例也有很大的区别。
在二、三碱基重复类型中占比例最高的碱基重复类型分别是AT/TA和AAG/TTC,在川西獐牙菜中二、三碱基重复类型中比例最高的碱基重复类型分别为AT/TA和AAT/TTA[21],但是在唐古特红景天(Rhodiolaalgida)中的二碱基重复类型中比例较高的碱基重复类型为AG/GA和TC/CT,三碱基重复类型中各个重复类型所占的比例较均匀[22]。蓝玉簪龙胆中二、三碱基重复类型比例较高的重复类型与红景天的差别比较大,而与川西獐牙菜的差别没那么大,这可能与不同植物之间的亲缘关系有关。对蓝玉簪龙胆的微卫星长度进行分析可以得出,在微卫星中重复单元的长度大小和重复次数成负相关,并且微卫星的总长度与重复单元的长度成正相关。
通过对蓝玉簪龙胆的SNP的分析发现,GP-F含有253 789个SNPs位点,平均跨度699 bp,GP-L含有SNPs 249 417个,平均跨度711 bp,这比青杨(Populuscathayana,1/29 332 bp)的分布密度要高,这可能与物种、测序数量、SNP分布的不均一性有关[23]。蓝玉簪龙胆的SNP位点在非编码区所占的比例要比在编码区的比例高一些,和以往的研究结果一致[24~25],可能是由于进化的原因和自然选择所致。蓝玉簪龙胆的SNP位点在编码区中同义变换所占的比例(48.63%和47.96%)要远远大于非同义变换的比例(0.08%和0.08%),可能是由于功能基因序列相对稳定所造成的。
综上所述,我们在高通量转录组测序的基础下,充分挖掘了蓝玉簪龙胆SSR、SNP信息,为构建蓝玉簪龙胆遗传学图谱以及谱系地理学研究提供了多方面的分子基础;该研究所获得SSR以及SNP分子标记,还为以后龙胆属植物的生物多样性研究以及保护生物学的研究提供了基础。
1.Zheng D.The system of physico-geographical regions of the Qinghai-Xizang(Tibet) Plateau[J].Science in China(Series D),1996,39(4):410-417.
2.杨红澎,确生,吴锡冬,等.蓝玉簪龙胆中苷类成分的研究[J].中国中药杂志,2008,33(21):2505-2507.
Yang H P,Que S,Wu X D,et al.Studies on glycosides fromGentianaveitchiorum[J].China Journal of Chinese Materia Medica,2008,33(21):2505-2507.
3.Faver A,Yuan Y M,Küpfer P,et al.Phylogeny of subtribeGentianinae(Gentianaceae):biogeographic inferences despite limitations in temporal calibration points[J].Taxon,2010,59(6):1701-1711.
4.Zhang Z F,Yuan L,Lu L Y,et al.Hepatoprotective activity ofGentianaveitchiorumHemsl.against carbon tetrachloride-induced hepatotoxicity in mice[J].Chinese Journal of Natural Medicines,2014,12(7):488-494.
5.Mishiba K I,Yamane K,Nakatsuka T,et al.Genetic relationships in the genusGentianabased on chloroplast DNA sequence data and nuclear DNA content[J].Breeding Science,2009,59(2):119-127.
6.Feng S,Tang M C,Wang D M.New evidence for the Qinghai-Xizang(Tibet) Plateau as a pilot region of climatic fluctuation in China[J].Chinese Science Bulletin,1998,43(20):1745-1749.
7.蔡英,李栋梁,汤懋苍,等.青藏高原近50年来气温的年代际变化[J].高原气象,2003,22(5):464-470.
Cai Y,Li D L,Tang M C,et al.Decadal temperature changes over Qinghai-Xizang Plateau in recent 50 years[J].Plateau Meteorology,2003,22(5):464-470.
8.Piao S L,Ciais P,Huang Y,et al.The impacts of climate change on water resources and agriculture in China[J].Nature,2010,467(7311):43-51.
9.Ellegren H.Microsatellites:simple sequences with complex evolution[J].Nature Reviews Genetics,2004,5(6):435-445.
10.Goldstein D B,Schlotterer C.Microsatellites:evolution and applications[M].Oxford,Enland:Oxford University Press,1999.
11.Wright J M,Bentzen P.Microsatellites:genetic markers for the future[M].Carvalho G R,Pitcher T J.Molecular genetics in fisheries.Netherlands:Springer,1995:117-121.
12.Zane L,Bargelloni L,Patarnello T.Strategies for microsatellite isolation:a review[J].Molecular Ecology,2002,11(1):1-16.
13.Vignal A,Milan D,Sancristobal M,et al.A review on SNP and other types of molecular markers and their use in animal genetics[J].Genetics Selection Evolution,2002,34(3):275-305.
14.Morin P A,Luikart G,Wayne R K,et al.SNPs in ecology,evolution and conservation[J].Trends in Ecology & Evolution,2004,19(4):208-216.
15.Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nature Biotechnology,2011,29(7):644-652.
16.Li S X,Yin T M.Map and analysis of microsatellites in the genome ofPopulus:the first sequenced perennial plant[J].Science in China Series C:Life Sciences,2007,50(5):690-699.
17.Mckenna A,Hanna M,Banks E,et al.The genome analysis toolkit:a Map Reduce framework for analyzing next-generation DNA sequencing data[J].Genome Research,2010,20(9):1297-1303.
18.Erlich Y,Mitra P P,Delabastide M,et al.Alta-Cyclic:a self-optimizing base caller for next-generation sequencing[J].Nature Methods,2008,5(8):679-682.
19.Jiang L H,Schlesinger F,Davis C A,et al.Synthetic spike-in standards for RNA-seq experiments[J].Genome Research,2011,21(9):1543-1551.
20.Brookes A J.The essence of SNPs[J].Gene,1999,234(2):177-186.
21.刘越,岳春江,王翊,等.藏茵陈川西獐牙菜转录组SSR信息分析[J].中国中药杂志,2015,40(11):2068-2076.
Liu Y,Yue C J,Wang Y,et al.Data mining of simple sequence repeats in transcriptome sequences of Tibetan medicinal plant ZangyinchenSwertiamussotii[J].China Journal of Chinese Materia Medica,2015,40(11):2068-2076.
22.雷淑芸,高庆波,付鹏程,等.基于Solexa高通量测序的唐古特红景天(Rhodiolaalgida)微卫星信息分析[J].植物研究,2014,34(6):829-834.
Lei S Y,Gao Q B,Fu P C,et al.Analysis on Microsatellites inRhodiolaalgidaBased on Solexa Sequencing[J].Bulletin of Botanical Research,2014,34(6):829-834.
23.雷淑云,张发起,Gulzar K,等.利用高通量测序分析青藏高原地区青杨的SSR和SNP特征[J].林业科学研究,2015,28(1):37-43.
Lei S Y,Zhang F Q,Khan G,et al.Characteristic analysis of SSR and SNP inPopuluscathayanaon Qinghai-Tibetan Plateau by High-Throughput Sequencing[J].Forest Research,2015,28(1):37-43.
24.Lijavetzky D,Cabezas J,Ibáez A,et al.High throughput SNP discovery and genotyping in grapevine(VitisviniferaL.) by combining a re-sequencing approach and SNPlex technology[J].BMC Genomics,2007,8(1):424-435.
25.An C F,Saha S,Jenkins J N,et al.Cotton(Gossypiumspp.) R2R3-MYB transcription factors SNP identification,phylogenomic characterization,chromosome localization,and linkage mapping[J].Theoretical and Applied Genetics,2008,116(7):1015-1026.
National Natural Science Foundation of China(31400322);International S&T cooperation projects of Qinghai Province(2014-HZ-812)
introduction:TIAN Zun-Zhe(1989—),male,master degree,mainly engaged in the research of plant genetic diversity.
date:2016-03-31
CharacteristicsofSSRandSNPinGentianaveitchioruminQinghai-TibetanPlateau,byHigh-throughputSequencing
TIAN Zun-Zhe1,2GAO Qing-Bo1CHEN Shi-Long1ZHANG Fa-Qi1*
(1.Key laboratory of Adaption and Evolution of Plateau Biota,Northwest Institute of Plateau Biology,Chinese Academy of Sciences,Xining 810001;2.University of Chinese Academy of Sciences,Beijing 100039)
We used the next generation sequencing(NGS) to capture the SSR and SNP marker inGentianaveitchiorum, sampled in Zeku, Qinghai Province. A total of 8588 simple sequence repeats were generated through lumina HiSeqTM2500. The dinucletide repeats were the highest(54.7%), followed by the trinucleotide repeats(41.3%). A proportion of tetranucletide, pentanucleotide and hexanucleotide repeats was less, which is 4% in total. In dinucleotide repeats, AT/TA repeats were the dominated ones, 9.85% and 9.5%, respectively. The length of nucleotide repeat and number of repeat were negative correlation, while there was a positive correlation between the total length of SSR sequences and the length of nucleotide repeat. A total of 253 789 and 249 417 SNPs were indenfied in GP-F and GP-L ofG.veitchiorum. The proportion of the SNPs located in the noncoding region were 51.29% and 51.96%, respectively. The proportion of synonymous transition(48.63% and 47.96%) in coding region was significantly higher than that of nonsynonymous transition(0.08% and 0.08%), which might be caused by relatively conservative domains of functional genes.
Gentianaveitchiorum;next generation sequencing;SSR;SNP;Qinghai-Tibetan Plateau
国家自然科学基金(31400322);青海省国际科技合作项目(2014-HZ-812)
田尊哲(1989—),男,硕士研究生,主要从事植物遗传多样性学研究。
* 通信作者:E-mail:fqzhang@nwipb.cas.cn
2016-03-31
* Corresponding author:E-mail:fqzhang@nwipb.cas.cn
Q949.776.4
A
10.7525/j.issn.1673-5102.2016.05.016
猜你喜欢
基层中医药(2022年7期)2022-11-17
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
知识就是力量(2022年2期)2022-03-01
天然产物研究与开发(2018年8期)2018-09-10
中成药(2017年12期)2018-01-19
四川动物(2017年6期)2017-12-12
四川动物(2017年4期)2017-07-31
中成药(2017年3期)2017-05-17
种子(2016年9期)2016-12-04
中成药(2016年4期)2016-05-17
