
MoP(001)表面苯胺C―N键断裂机理
2016-11-08 06:00李邵仁鲁效庆朱后禹郭文跃中国石油大学华东理学院山东青岛266580
物理化学学报 2016年2期
李邵仁 鲁效庆 朱后禹 郭文跃(中国石油大学(华东)理学院,山东青岛266580)
MoP(001)表面苯胺C―N键断裂机理
李邵仁鲁效庆*朱后禹郭文跃
(中国石油大学(华东)理学院,山东青岛266580)
油品脱氮可以减少燃烧过程中氮氧化物的排放,并且减弱催化剂的中毒现象,过渡金属磷化物因具有高催化活性和优异的稳定性而成为最具应用前景的新型加氢脱氮(HDN)催化剂。本工作基于周期性平板模型,通过密度泛函理论(DFT)计算研究了苯胺在磷化钼(MoP)(001)表面的吸附以及C―N断键机理。结果表明在MoP(001)表面,苯胺吸附以平躺吸附构型为主,有较大的吸附能,C―C键和C―N键均被吸附活化;苯胺C―N键的直接断裂路径主要起始于与共吸附H2发生反应,产物为苯和氨,吸附的环己胺的C―N键断裂主要路径是环己胺与共吸附的H发生反应脱去氨基,生成产物为环己烯和氨。
磷化钼;苯胺;加氢脱氮;碳氮键;密度泛函理论
doi:10.3866/PKU.WHXB201511242
最近,一种新型的过渡金属磷化物在加氢精制反应中被发现拥有很高的活性和稳定性2-4。此外,过渡金属磷化物还展现出非常优良的化学和物理性质,比如:它们是良好的电导体和热导体,具有超高硬度和高熔点5。在这些磷化物中,MoP引起了人们广泛的关注6-9。目前,已经有一些研究MoP催化剂表面上HDN反应的实验文献。实验研究6,10表明负载型和无负载MoP固有的HDN活性都比商用钼基催化剂高很多。更重要的是,MoP催化剂的HDN活性在含硫化合物存在的加氢精制反应中能保持稳定8,11。例如,Oyama等8报道在喹啉HDN和二苯并噻吩HDS反应同时进行时,相比商用催化剂氧化铝负载的MoP表现出更高和更稳定的活性。MoP由于其高的稳定性和合适的活性而被认为是HDN反应中最有前景的加氢精制催化剂8,12,13。然而,这些工作的重点在于催化剂的制备和HDN活性以及催化表面性质的表征上,MoP催化表面的HDN机理的细节尚不完全明确,需要机理性的研究提供相关基元反应的能量和结构信息,从原子水平上认清催化剂表面的性质。
随着实验技术和手段的进步,关于苯胺类化合物在磷化物催化剂表面的HDN机理引起了广泛关注。部分研究14报道认为苯胺类化合物HDN反应机理是苯胺类化合物通过加氢反应生成环己胺类化合物,然后通过β-H消去机理生成环己烯类化合物;但也有观点15认为环己胺类化合物可以通过SH取代NH2的亲核取代反应而脱氮。此外,极少量的苯胺类化合物是通过直接氢解生成苯类和氨基16。尽管上述研究对苯胺类化合物C―N键断裂机理进行了相关的阐述,但受限于实验条件的差异以及研究对象的不同,目前无法得到一致的结论。更重要的是,这些含氮化合物在催化剂表面到底如何进行HDN反应?相关基元反应步骤的微观信息如何?由于实验研究的局限性以及侧重点不同,上述问题一直无法解决。
为深入认识HDN反应机理,我们利用密度泛函理论(DFT)从原子水平进行了相关的研究。在本文中,我们选取苯胺类化合物中较小的分子——苯胺作为研究对象,研究其在MoP(001)表面C―N键的断裂。并考虑实际条件下,催化剂表面H2、H2S的存在会对C―N键的断裂起着特殊作用,如在MoP表面上发生的HDN反应中H2S会促进邻甲基苯胺分子的C―N键的断裂10。因此,本文从理论上研究催化剂表面有/无H2、H、SH和S情况下苯胺在MoP(001)表面C―N键的断裂机理,反应初态与末态均选取吸附物最稳定吸附位置,考虑到H在MoP(001)表面fcc和hcp位置可以相互扩散17,因此我们在反应中考虑了两个位置上H参与反应的情况。通过比较各反应的能垒,提出最优反应路径,从理论上揭示了MoP表面上苯胺C―N键的断裂机理,为改进MoP催化剂表面HDN反应活性提供一定的理论依据。
2 计算方法和模型
本文所有量子化学计算都在Materials Studio软件包中的Dmol3模块18-20完成。计算参数选择如下:交换关联近似方法选用广义梯度近似(GGA)和PW91泛函相结合的方法(GGA-PW91)21,22;基组选用双数值型基组加极化函数(DNP);核赝势选用密度泛函半核芯赝势(DSPP);自旋处理选用自旋极化(spin unrestricted);计算中选用medium网格质量。其中能量、梯度、最大位移和自洽场(SCF)的收敛标准分别为2´10-5hartree、4´10-2hartree∙nm-1、5´10-4nm和1´10-5hartree。单点能计算为校正MoP(001)表面和吸附质的电荷,核赝势选用全电子(all electron)23。布里渊区积分选用2´2´1的Monkhorst-Pack k点24。利用该计算参数和收敛标准计算的体相MoP晶格常数为a=b=0.3252 nm和c=0.3216 nm,这与实验值(0.3223和0.3191 nm)25和理论值(0.3218和0.3176 nm)26基本一致。利用完全线性同步(LST)和二次同步变换(QST)方法27搜索过渡态(TS),同时使用振动频率分析并确认过渡态。
本文计算吸附能(Eads)公式为:其中,Eadsorbate是气相吸附质分子的能量,EM是吸附剂体系的能量,Eadsorbate/M是吸附质吸附在吸附剂表面整个体系的能量。因此,正的吸附能越大代表吸附越稳定。计算吸附剂表面基元反应的反应能(DE)和能垒(Ea)公式为:DE=EFS/M-EIS/M和Ea=ETS/M-EIS/M,其中EIS/M,ETS/M和EFS/M分别为吸附剂表面上反应初态(IS)、过渡态以及反应末态(FS)体系的总能量。前期理论28,29和实验6研究表明MoP催化剂表面Mo暴露面具有较高的稳定性和催化活性,有利于吸附质的表面吸附。选取了与我们前一篇文章17同样的基底,如图1所示,Mo终端的MoP (001)表面包括三层Mo与二层P,共45个原子,超晶胞真空层为1.3nm。计算过程中,固定MoP(001)表面底下两层原子与计算的体相点阵位置,上面三层原子与吸附质采取无对称约束的优化。
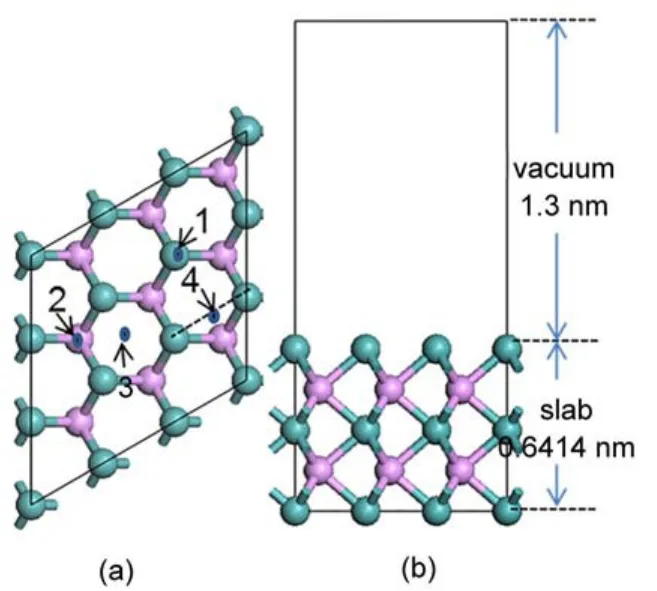
图1Mo终端的MoP(001)表面结构模型Fig.1 Structure model of the Mo-terminated MoP(001)surface
3 结果与讨论
3.1苯胺及环己胺的吸附
苯胺HDN有两条反应路径,如图2所示:(1)苯胺通过直接断裂C―N键形成苯;(2)苯胺通过加氢形成环己胺,然后再催化断裂C―N键形成环己烯。由于不饱和含氮化合物的加氢反应优先于C―N键的断裂是HDN反应中最基本的步骤30,并且我们研究的侧重点在于C―N键的断裂机理、反应路径以及相关基元步骤的能量、结构等信息。因此我们这里只探讨苯胺以及其加氢饱和产物环己胺C―N键的断裂机理,至于反应网络中加氢过程没有考虑。苯胺及其加氢饱和产物环己胺在MoP(001)表面的稳定吸附构型见图3,表1列出了相关的吸附能及重要的结构参数。如图3所示,在MoP(001)表面苯胺形成了三种稳定吸附构型(图3 (a,b,c)),分别定义为fcc-top、hcp-top和top-top构型(前者指代苯环的吸附位,后者指代氨基吸附位)。由于苯胺中的苯环与氨基均能与表面相互作用,吸附以平躺吸附构型为主,这类吸附构型对应的吸附能较大,其中最稳定构型为hcp-top构型(吸附能为2.63 eV)。在最稳定构型中,N―Mo键长为0.2289 nm,C―N键长为0.1442 nm,最短的C―Mo键长为0.2245 nm。环己胺因饱和碳环与表面存在较强排斥作用,只有氨基与表面直接相互作用,两种稳定吸附构型对应的吸附能都相对较小,分别为0.88 eV(hcp-top)和0.89 eV(fcc-top)。
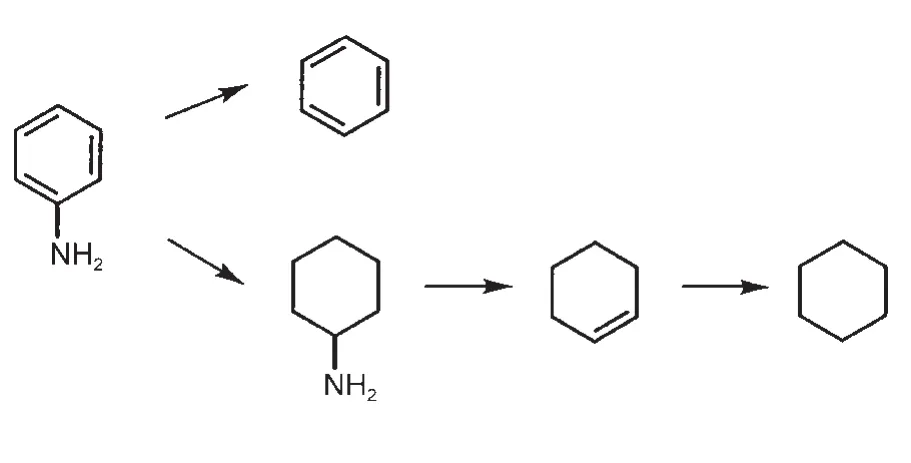
图2 苯胺HDN反应网络Fig.2 HDN reaction network of aniline
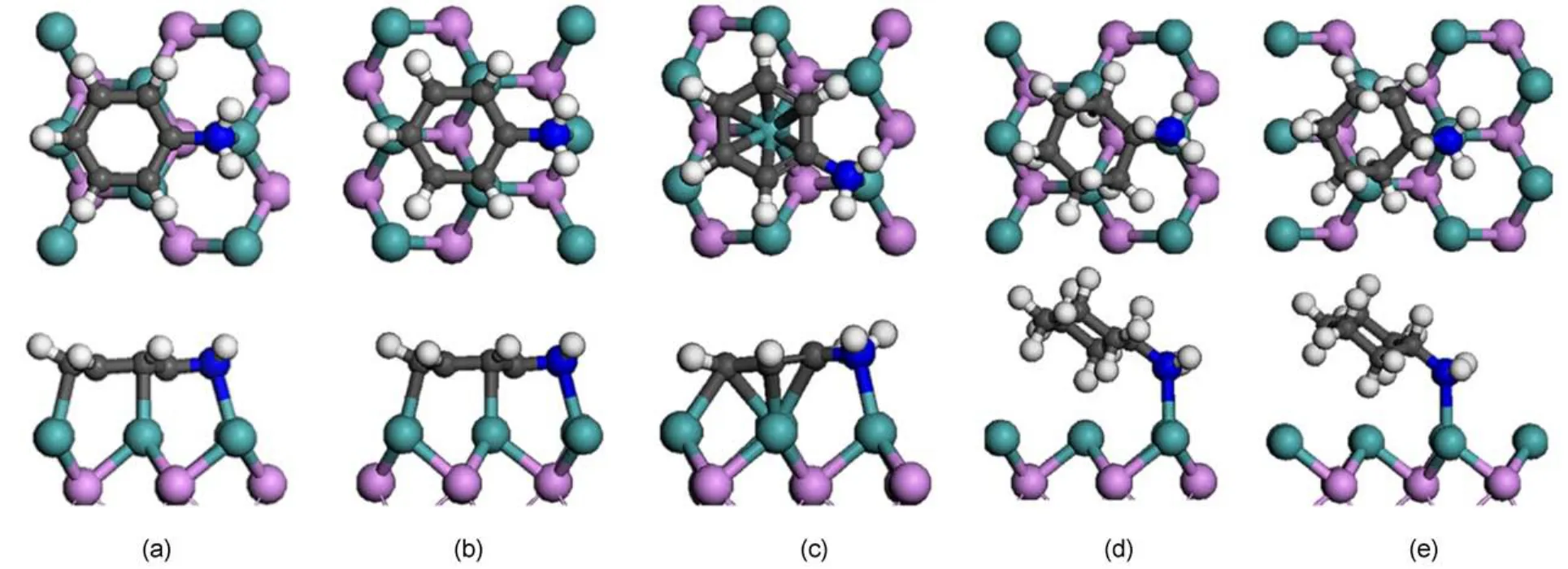
图3 苯胺及环己胺在MoP(001)表面稳定吸附构型的俯视图和侧视图Fig.3 Top and side views of the stable adsorption structures of aniline and cyclohexylamine on MoP(001)surface
对比苯胺与环己胺各自在吸附前后C―N键的变化情况(见表1),可以看出苯胺C(sp2)―N键活化程度比环己胺C(sp3)―N键强。这是因为苯胺从催化剂表面接受电子,苯环与N都与表面发生较强的相互作用,而环己胺通过N向表面转移电子,与表面相互作用较弱。尽管如此,在催化剂表面苯胺最稳定吸附构型中C(sp2)―N键比环己胺最稳定吸附构型中C(sp3)―N键还是短(0.1442 nm vs 0.1494 nm)。不过,苯胺中C―C键长在吸附前后变化较明显,表明芳香环因吸附被表面活化,这对苯环加氢有利。
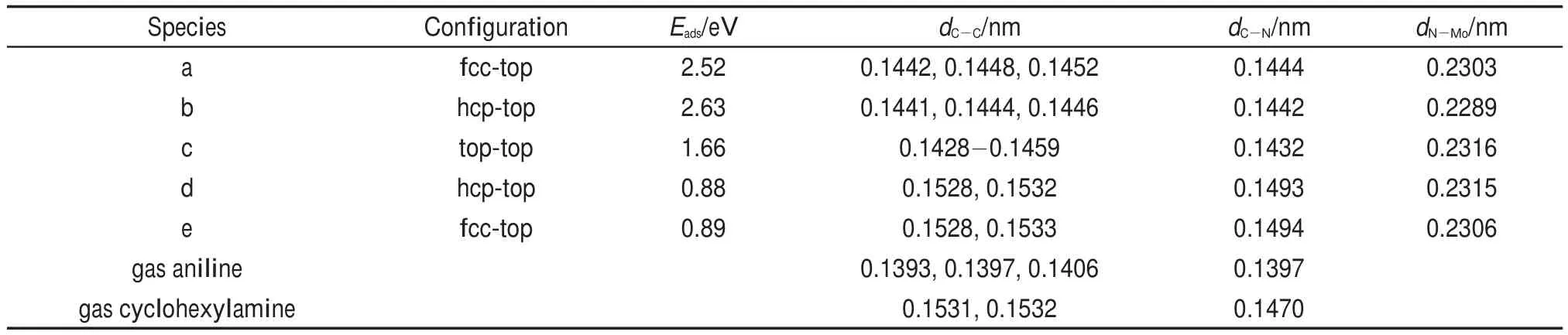
表1 苯胺和环己胺在MoP(001)表面的吸附构型、吸附能(Eads)以及结构参数Table 1 Adsorption configurations,adsorption energies(Eads),and structural parameters for aniline and cyclohexylamine on MoP(001)surface
3.2苯胺C― N键的断裂机理
3.2.1苯胺
苯胺及其加氢饱和产物环己胺C―N键的断裂所有可能基元反应的Ea和ΔE值见表2。对于苯胺C―N键的断裂机理,首先计算了苯胺C―N键直接断裂,相应的反应路径见图4(a)。如图4(a)所示苯胺分解生成苯基和氨基反应,C―N键直接断裂反应开始于苯胺在MoP(001)表面hcp-top吸附,C―N键在Mo参与下发生断裂而到达TS1。在TS1中,C―N键已经断裂,C与N之间距离为0.2174 nm,新的Mo―C键已经形成,Mo与C距离为0.2371 nm,Mo―N键拉长到0.2080 nm。TS1之后,苯基向邻近hcp位移动,氨基向邻近桥位移动最终到达反应末态。该过程接近热中性(反应仅放热0.04 eV),计算的能垒为2.07 eV。产物苯基和氨基的加氢过程,见Supporting Information中图S1 (a)和(b),反应初态均为各自稳定构型与邻近hcp位H的共吸附构型,反应物借助中间Mo相互靠近并逐渐成键,最终形成桥位吸附的苯或顶位吸附的氨气。两步反应的能垒分别为0.77和2.02 eV,对应的反应热为-0.03和0.55 eV。该结果说明若苯胺C―N键发生直接断裂,形成的苯基很容易加氢形成苯,而氨气的形成则比较困难。除此之外,我们还考虑了催化剂表面吸附的H作用下C―N键断裂的情况(见图4(b))。在过渡态TS2中,C―N键已断裂,相应的距离为0.2216 nm,氨基位于Mo顶位,Mo―N键长为0.2173 nm,N与H之间的距离缩短至0.1786 nm。该反应由于很高的能垒(3.20 eV)无法实现。H2分子同时攻击苯环与氨基的氢解反应(见图4(c))对应1.73 eV的能垒和0.05 eV的反应热,表明该反应是有可能发生的。在TS3中,C―N键已断裂并且氨气分子已形成,解离的H位于Mo顶位。总之,氨基在苯环外,C―N键具有直接断裂的可能性,但苯环π电子与氨基N上的孤对电子存在共轭效应,C―N键断裂能垒较高。
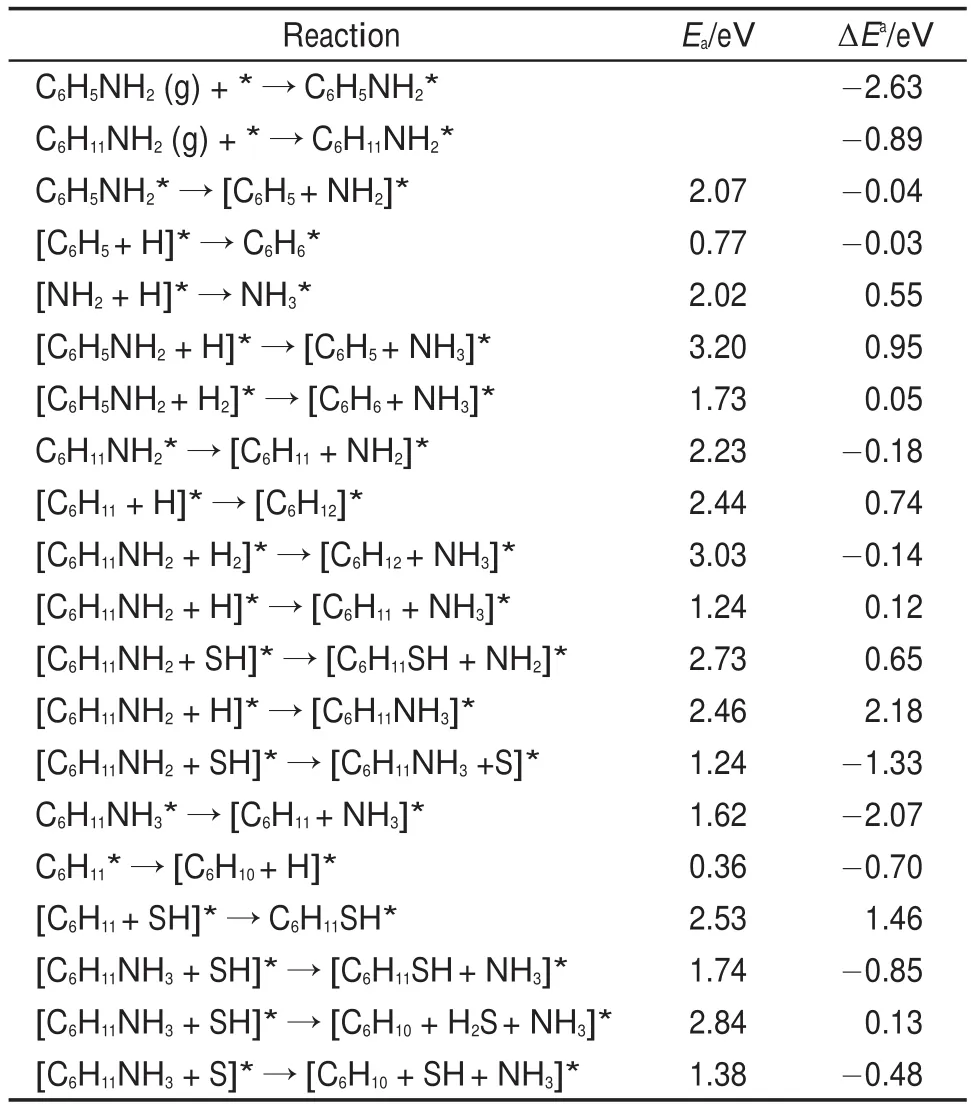
表2 苯胺和环己胺在MoP(001)表面C―N键断裂所涉及可能基元反应的能垒(Ea)和反应能(ΔE)Table 2 Calculated energy barriers(Ea)and reaction energies(ΔE)of possible elementary reactions involved in C―N bond cleavage of aniline and cyclohexylamine on MoP(001)surface
3.2.2环己胺
对于环己胺C―N键的断裂,由于环己胺C(sp3)―N键与苯胺C(sp2)―N键中Cα饱和差异,决定了其断裂的难易。首先图5(a)显示了环己胺C―N键的直接断裂细节情况。计算表明该反应能垒为2.23 eV,反应放热0.18 eV,与苯胺C―N键断裂情况相比,能垒稍稍高出0.16 eV。分析其原因,虽然环己胺C―N键比苯胺的长,但环己胺与表面的相互作用很弱,并且环己基与氨基在Mo顶位发生竞争吸附也会导致反应能垒增高。该反应产物环己基加氢反应(如图S1(c)所示)的能垒为2.45 eV。与苯胺一样,我们还考虑了环己胺在催化剂表面共吸附的H2、H以及SH作用下C―N键断裂的情况(见图5(b,c,d))。如图5(b)所示H2与环己胺共吸附在MoP(001)表面,经历TS5,C―N键断裂生成氨气和环己烷,该反应能垒较高为3.03 eV,从动力学角度分析,这个反应很难进行。图5(c)所示以环己胺与H在MoP(001)表面共吸附为初态的反应,经过TS6生成环己基和氨气。在TS6中环己胺C―N键已断裂,C与N距离0.2216 nm,H靠近氨基中N,两原子距离0.1786 nm表明N与H还未成键。该过程反应能垒为1.24 eV,反应需要吸收热量0.12 eV,适当的升高温度有利于该反应的进行。对于环己胺与SH取代反应,如图5(d)所示,以环己胺与SH在MoP(001)表面共吸附为初态的反应,经历TS7,SH取代环己胺上的NH2,生成环己硫醇。这步反应需要克服能垒2.73 eV,吸收热量0.65 eV。
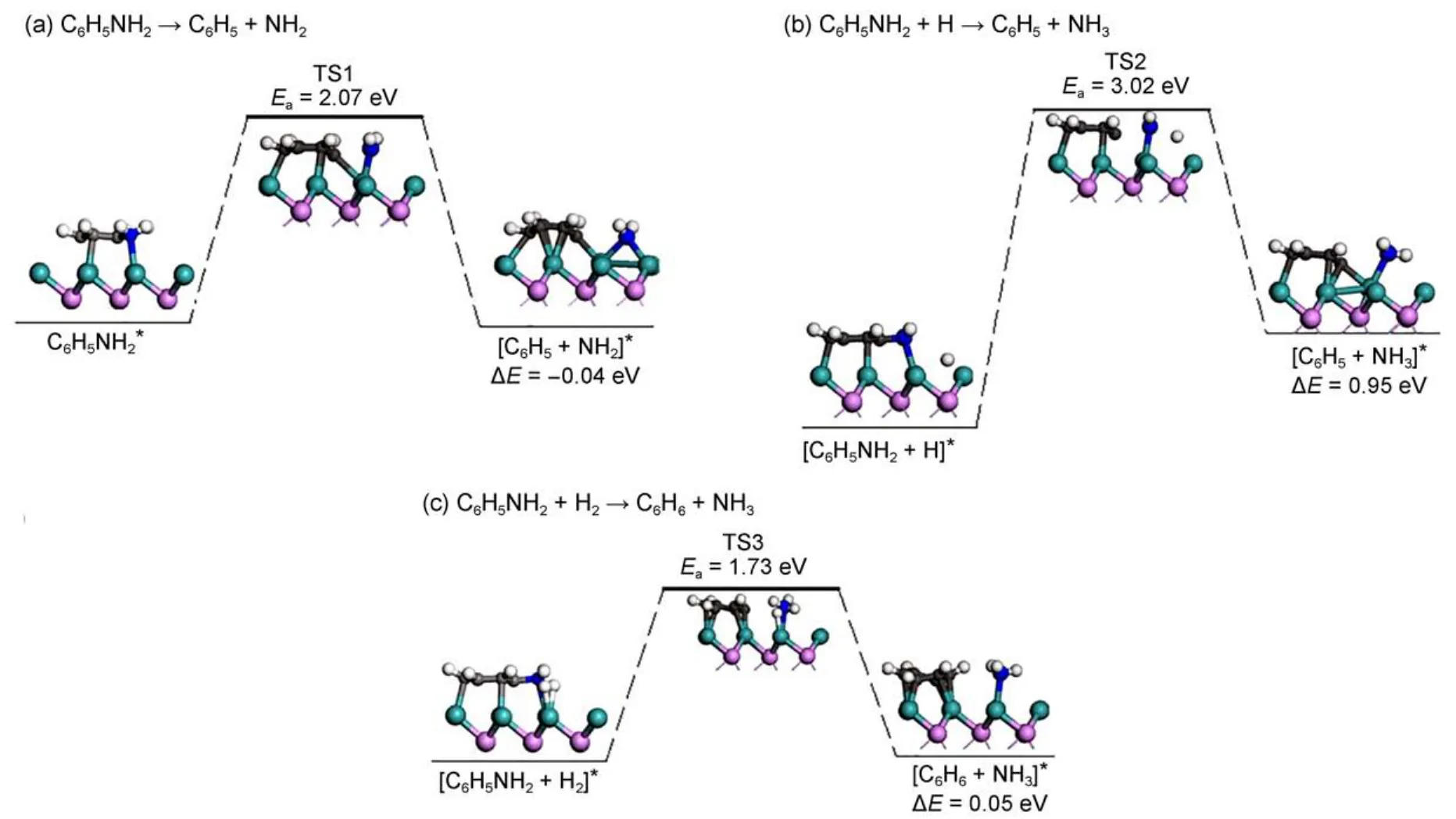
图4 苯胺在MoP(001)表面C ―N键直接断裂、氢解反应Fig.4 C ―N bond direct cleavage and hydrogenolysis reactions of aniline on MoP(001)surface
环己胺C―N键断裂可能的断裂机理还有消去和亲核取代机理。这两种机理环己胺首先都要通过N的孤对电子与质子结合,即发生质子化作用形成一个容易脱去的季铵基团。对于环己胺的质子化反应中氢质子来源首先考虑了催化剂表面H。此质子化反应反应是从H与环己胺共吸附开始的(见图5(e)),随着反应的进行,经过TS8,H与N靠近最终键合成N―H键,这步反应能垒高达2.46 eV,而且需要吸收热量2.18 eV,这从动力学和热力学角度都不利于该反应进行。除了表面H外,Brønsted酸(B酸)活性中心也可以提供质子,由于SH具有质子性质的氢原子,而且SH很低的解离能垒对这种加氢反应也有积极的作用17,所以SH是最可能的B酸性中心。对于环己胺的质子化反应发生在存在SH的催化剂表面,如图5(f)所示。在这个反应中,环己胺向表面靠近的过程中通过N的孤对电子与SH上的H结合完成质子化,而S始终占据最初的位置。在TS9中,S―H键已断裂,相应的距离为0.2083 nm,N与H之间的距离为0.2360 nm。该反应的能垒为1.24 eV,反应能为-1.33 eV。
环己胺完成质子化,将有两种可能的C―N键断裂路径:消去反应(E1、E2消去反应)和亲核取代反应(SN1、SN2取代反应)。当反应中不存在SH时,催化表面仅覆盖H,因此无酸碱性活性位和亲核试剂存在,C―N键只能通过E1消去机理断裂。E1消去机理包括两步反应(见图6(a,b))。第一步质子化的环己胺的脱氨过程,C―N键在桥位附近断裂,形成fcc位吸附的环己基和顶位吸附的氨气。在TS10中,C与N之间的距离由反应初态的0.1516 nm延长到0.2607 nm。该反应具有较合理的能垒(1.62 eV)和高放热性(反应能为-2.07 eV)。质子化的环己胺脱氨后,氨气在表面脱附。第二步以吸附在fcc位的环己基为初态,环己基的Cβ―H键被与Hβ成键的Mo活化而发生断裂,脱去的H吸附在邻近fcc位。在TS11中,Cβ―H键已断裂,相应的距离为0.1644 nm;一个新的Mo―Cβ键形成,键长为0.2385 nm;Mo―H键长由反应初态时的0.1926 nm变为0.1794 nm。该过程包含0.36 eV的能垒和-0.70 eV的反应能,表明E1消去反应的第二步很容易进行。当SH也存在于反应中时,由于亲核取代机理中,SH是亲核试剂,此时催化表面存在酸碱性活性位,消去和取代机理都可能发生。SN1机理包括如图6(a,c)所示两步反应,第一步反应与上面提到的E1消去机理第一步反应相同,第二步反应环己基与邻近fcc位的SH相互靠近,最终形成环己硫醇。在TS12中,S已移至桥位,相应的两个Mo―S键长为0.2403和0.2435 nm;S与Cα之间的距离为0.3038 nm。由于该反应对应的能垒为2.53 eV并且反应需要吸热1.46 eV,所以SN1机理反应很难发生。对于SN2机理(见图6(d)),质子化的环己胺和SH共吸附在催化剂表面为反应初态,质子化的环己胺C―N键受到SH影响,然后发生断裂,最终形成吸附在Mo顶位上的NH3和通过S与催化剂表面相互作用的环己醇。在TS13中,C和N距离为0.2580 nm表明C―N键已断裂。此反应过程中能垒为1.74 eV,反应热为-0.85 eV。对于E2消去反应,我们考虑了表面存在SH或者S的反应(如图6(e,f))。图6(e)所示的是质子化的环己胺与SH共吸附作为反应初态,最终生成环己烯、硫化氢和氨;图6(f)所示质子化的环己胺与邻近fcc位吸附的S作为反应初态,强碱S通过它的孤对电子攫取一个Hβ,Cα―Cβ键的收缩从而导致Cα―N键的断裂。在TS14中,Cβ―H、Cα―N键均已断裂,对应的距离分别为0.3494和0.3055 nm;同时Cβ与Cα之间π键形成,Cβ与Cα之间距离为0.1327 nm,S与脱去的Hβ之间的距离为0.1485 nm。两者反应能垒分别为2.84和1.38 eV,反应热分为0.13和-0.48 eV,从动力学和热力学角度对比两反应,质子化的环己胺与强碱S共吸附反应更容易发生。
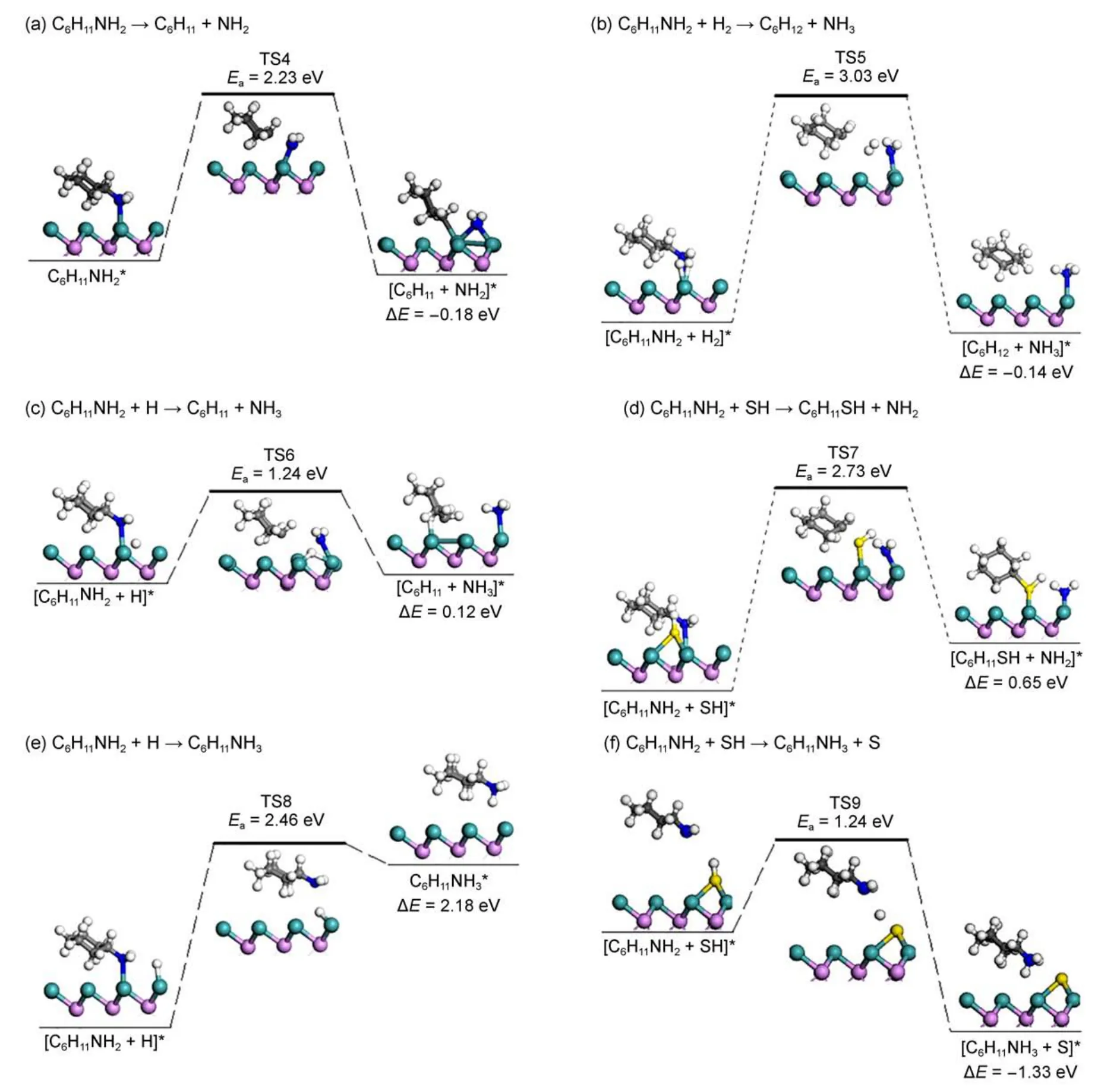
图5 环己胺在MoP(001)表面C ―N键直接断裂、氢解反应以及质子化反应Fig.5 C ―N bond direct cleavage,hydrogenolysis and quaternization reactions of cyclohexylamine on MoP(001)surface
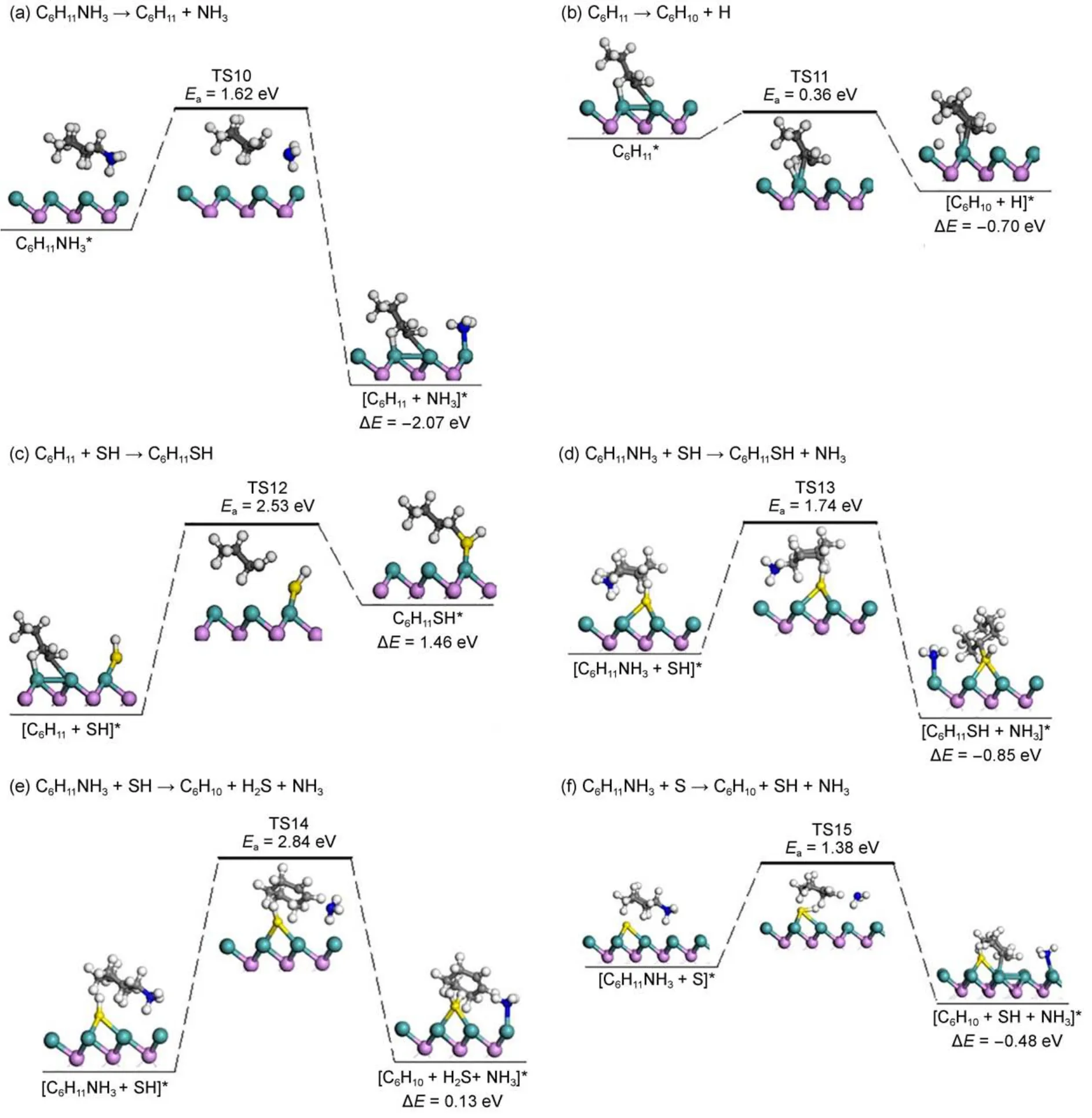
图6 环已胺C ―N键断裂的E1、SN1、E2、SN2反应路径Fig.6 E1,SN1,E2,and SN2 reaction paths of C ―N bond cleavage of cyclohexylamine
3.2.3苯胺与环己胺C―N键断裂机理总结
图7为苯胺C―N键断裂机理示意图,其中包括每步反应的能垒和反应能。通过图7可以看出苯胺C―N键直接断裂的能垒(2.07 eV)很高,反应较难发生,但受到表面共吸附H2影响下,苯胺C―N键断裂反应能垒有所降低(1.73 eV),这也使得很少部分苯胺通过氢解反应可以直接脱氨基而形成苯,这点与相关苯胺类HDN实验中有检测到苯的形成的报道16一致。苯胺加氢饱和产物环己胺C―N键直接断裂能垒也很高(2.23 eV),不过受到表面共吸附物种H影响下,环己胺C―N键断裂能垒下降很多,仅为1.24 eV,从动力学角度看这个反应的能垒是较合理的,反应较容易发生,从热力学角度,该反应也仅需要吸收很少热量(0.12 eV)。如考虑到催化剂表面吸附有SH存在,环己胺发生质子化反应能垒同样为1.24 eV,该反应为放热反应(-1.33 eV),与上面H攻击C―N键脱氨基反应对比,从热力学角度,该反应更容易进行。环己胺与表面吸附H发生质子化反应,能垒高达2.46 eV,且需要吸收热量2.18 eV,无论动力学还是热力学角度该反应都很难正向进行。但是该反应逆向反应,即质子化的环己胺N―H断裂,能垒仅为0.28 eV,且为放热反应。对比质子化的环己胺后续的C―N键断裂各反应能垒(1.38-2.84 eV),质子化环己胺N―H断裂反应进行的可能性最大。这说明即使环己胺在催化剂MoP(001)表面与共吸附SH完成质子化反应,但生成质子化的环己胺更倾向于在催化剂MoP(001)表面进行N―H断裂脱氢反应,又生成环己胺。因此,吸附的环己胺C―N键断裂最优路径为与表面H反应生成氨与环己基。环己基脱氢反应能垒很低(0.36 eV),且反应为放热反应,最终生成环己烯。考虑到环己胺与表面吸附能为0.89 eV,相比其C―N键断裂能垒都低,所以表面环己胺脱附相比其C―N键断裂更有利。
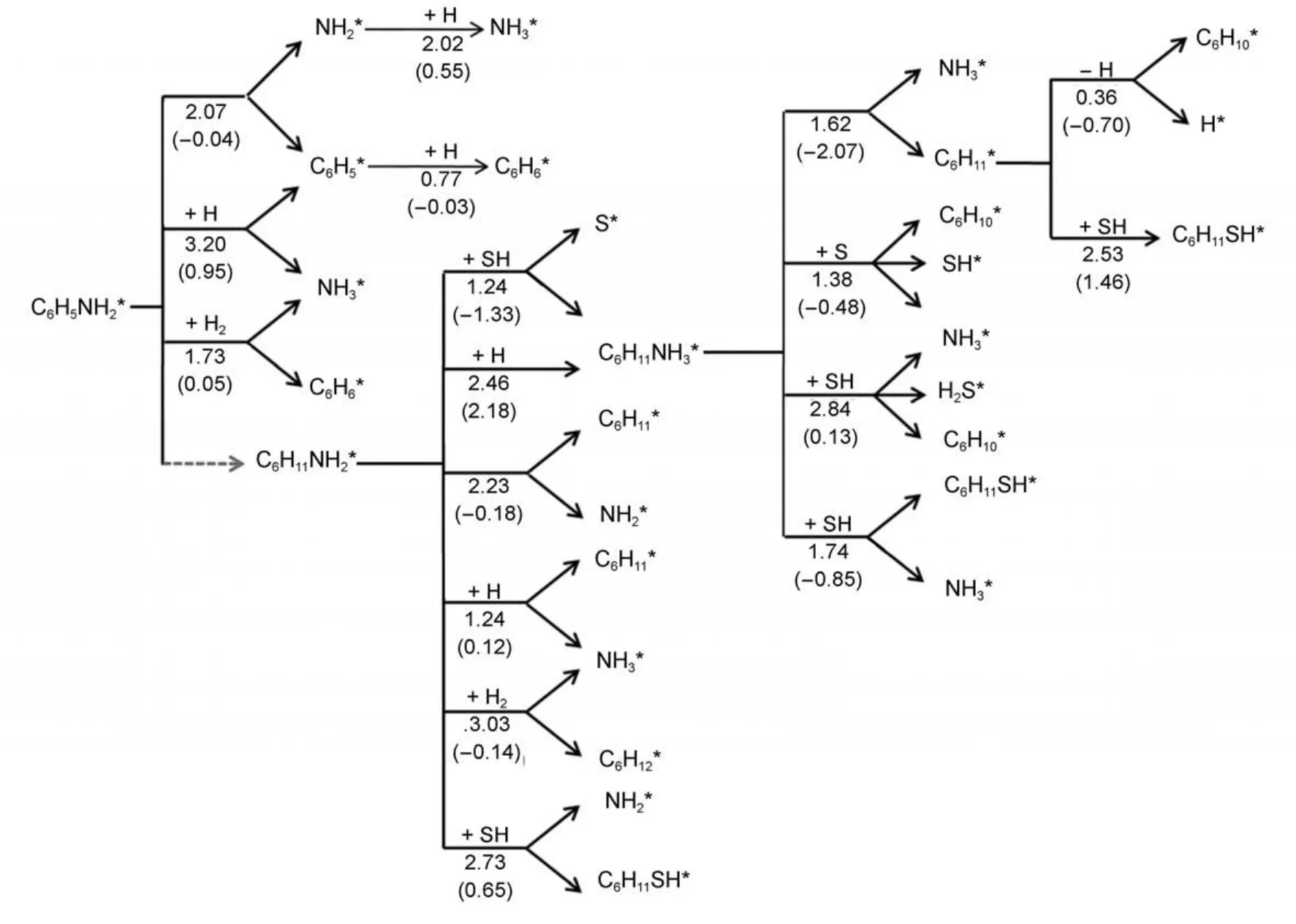
图7 苯胺在MoP(001)表面C―N键断裂机理示意图Fig.7 Reaction mechanism scheme of aniline C―N bond cleavage on MoP(001)surface
综上所述,苯胺C―N键直接断裂路径主要起始于与表面共吸附H2发生反应,生成物包含苯。而环己胺C―N键断裂主要路径是吸附的环己胺受到共吸附H攻击C―N键而脱氨基,最终产物中包含环己烯。该结论与实验结果6一致:研究发现苯胺或苯胺类化合物在MoP催化剂上HDN反应时,无论反应中是否加入H2S,HDN的主要产物均为环己烯。值得一提的是,苯胺通过氢解反应C―N键断裂形成的苯非常少,而且苯胺在催化剂表面的吸附比苯强,很难与苯胺竞争加氢,因此,反应网络中的环己稀是由苯胺脱氨形成,而非苯加氢形成31。
4 结论
利用密度泛函理论研究了苯胺和环己胺在MoP(001)表面吸附及其在HDN反应中C―N键的断裂机理,提供了相关基元反应的路径、结构和能量等信息。苯胺吸附以平躺吸附构型为主,吸附能范围1.66-2.63 eV,为较强的化学吸附,C―C键和C―N键均被活化,促进了脱氮反应的进行,而其饱和加氢产物环己胺最稳定结构吸附能0.89 eV,与表面作用相对而言要弱,它的脱附比C―N键断裂更有利。苯胺C―N键直接断裂路径主要起始于与共吸附H2发生反应,生成物包含苯。而其饱和加氢产物环己胺C―N键断裂主要路径是吸附的环己胺受到共吸附H攻击C―N键而脱除氨基,最终产物中包含环己烯。H2S的存在不影响环己胺C―N键断裂能垒,共吸附的H2和H有助于苯胺C―N键断裂。
SupportingInformation:Hydrogenation reactions of C6H5, NH2,and C6H11on MoP(001)have been included.This information is available free of charge via the internet at http://www. whxb.pku.edu.cn.
References
(1)Zeuthen,P.;Knudsen,K.G.;Whitehurst,D.D.Catal.Today 2001,65(2-4),307.doi:10.1016/S0920-5861(00)00566-6
(2)Phillips,D.C.;Sawhill,S.J.;Self,R.;Bussell,M.E.J.Catal. 2002,207(2),266.doi:10.1006/jcat.2002.3524
(3)Clark,P.;Wang,X.;Oyama,S.T.J.Catal.2002,207(2),256. doi:10.1006/jcat.2002.3520
(4)Oyama,S.T.;Wang,X.;Lee,Y.K.;Chun,W.J.J.Catal. 2004,221(2),263.doi:10.1016/S0021-9517(03)00017-4
(5)Oyama,S.T.J.Catal.2003,216(1-2),343.doi:10.1016/ S0021-9517(02)00069-6
(6)Stinner,C.;Prins,R.;Weber,T.J.Catal.2000,191(2),438. doi:10.1006/jcat.1999.2808
(7)Li,W.;Dhandapani,B.;Oyama,S.T.Chem.Lett.1998,168 (3),207.
(8)Oyama,S.T.;Clark,P.;Teixiera da Silva,V.L.S.;Lede,E.J.; Requejo,F.G.J.Phys.Chem.B 2001,105(21),4961.doi: 10.1021/jp004500q
(9)Lü,C.Q.;Ling,K.C.;Wang,G.C.Acta Phys.-Chim.Sin. 2009,25(11),2336.[吕存琴,凌开成,王贵昌.物理化学学报,2009,25(11),2336.]doi:10.3866/PKU.WHXB20091038
(10)Zuzaniuk,V.;Prins,R.J.Catal.2003,219(1),85.doi: 10.1016/S0021-9517(03)00149-0
(11)Sun,F.X.;Wu,W.C.;Wu,Z.L.;Guo,J.;Wei,Z.B.;Yang,Y. X.;Jiang,Z.X.;Tian,F.P.;Li,C.J.Catal.2004,228(2),298. doi:10.1016/j.jcat.2004.09.002
(12)Clark,P.;Oyama,S.T.J.Catal.2003,218(1),78.doi: 10.1016/S0021-9517(03)00086-1
(13)Rodriguez,J.A.;Kim,J.Y.;Hanson,J.C.;Sawhill,S.J.; Bussell,M.E.J.Phys.Chem.B 2003,107(26),6276.doi: 10.1021/jp022639q
(14)Geneste,P.;Moulinas,C.;Olivé,J.L.J.Catal.1987,105(1), 254.doi:10.1016/0021-9517(87)90025-X
(15)Rota,F.;Prins,R.J.Mol.Catal.A:Chem.2000,162(1-2), 367.doi:10.1016/S1381-1169(00)00303-4
(16)Mou,J.;Kapteijn,F.;Prins,R.J.Catal.1997,168(2),491. doi:10.1006/jcat.1997.1650
(17)Li,Y.;Guo,W.Y.;Zhu,H.Y.;Zhao,L.M.;Li,M.;Li,S.R.; Fu,D.L.;Lu,X.Q.;Shan,H.H.Langmuir 2012,28(6), 3129.doi:10.1021/la2051004
(18)Delley,B.J.Chem.Phys.1990,92(1),508.doi:10.1063/ 1.458452
(19)Delley,B.J.Chem.Phys.1996,100(15),6107 doi:10.1021/ jp952713n
(20)Delley,B.J.Chem.Phys.2000,113(18),7756.doi:10.1063/ 1.1316015
(21)Perdew,J.P.;Wang,Y.Phys.Rev.B 1986,33(12),8800.doi: 10.1103/PhysRevB.33.8800
(22)Perdew,J.P.;Wang,Y.Phys.Rev.B 1992,45(23),13244.doi: 10.1103/PhysRevB.45.13244
(23)Ren,J.;Huo,C.F.;Wen,X.D.;Cao,Z.;Wang,J.G.;Li,Y. W.;Jiao,H.J.J.Phys.Chem.B 2006,110(45),22563.doi: 10.1021/jp0640474
(24)Monkhorst,H.J.;Pack,J.D.Phys.Rev.B 1976,13(12),5188. doi:10.1103/PhysRevB.13.5188
(25)Rundgvist,S.;Lundström,T.Acta Chem.Scand.1962,17(1), 37.
(26)Winkler,B.;Knorr,K.;Hytha,M.;Milman,V.J.Phys.Chem. Solids 2003,64(3),405.doi:10.1016/S0022-3697(02)00293-7
(27)Halgren,T.A.;Lipscomb,W.N.Chem.Phys.Lett.1977,49 (2),225.doi:10.1016/0009-2614(77)80574-5
(28)Milman,V.;Winkler,B.;Gomperts,R.Chem.-Eur.J.2004,10 (24),6279.
(29)Liu,P.;Rodriguez,J.A.Catal.Lett.2003,91(3),247.
(30)Oyama,S.T.;Lee,Y.K.J.Phys.Chem.B 2005,109(6),2109 doi:10.1021/jp049194l
(31)Rota,F.;Prins,R.Stud.Surf.Sci.Catal.1999,127,319.doi: 10.1016/S0167-2991(99)80423-6
Mechanism of C―N Bond Cleavage in Aniline on MoP(001)Surface
LI Shao-RenLU Xiao-Qing*ZHU Hou-YuGUO Wen-Yue
(College of Science,China University of Petroleum,Qingdao 266580,Shandong Province,P.R.China)
Denitrogeneration of petroleum products can reduce NOxemission during combustion,and relieve the poisoning of catalysts.Because of their high catalytic activities and excellent stabilities,transition metal phosphides exhibit great potential as novel,promising hydrodenitrogeneration(HDN)catalysts.Based on a periodic slab model,we investigated the adsorption and C―N bond cleavage mechanism of aniline on MoP(001) surface by density functional theory(DFT)calculations.The results show that aniline adsorption prefers a flat configuration,with larger adsorption energies,in which the C―C and C―N bonds are activated.The direct C―N bond cleavage mechanism of aniline proceeds mainly via deamination with co-adsorbed H2,producing benzene and ammonia.The C―N bond cleavage mechanism for adsorbed cyclohexylamine proceeds via deamination,with co-adsorbed H,and the main products are cyclohexene and ammonia.
Molybdenum phosphide;Aniline;Hydrodenitrogeneration;C―N bond; Density functional theory
1 引言
油品中的含氮化合物不仅在燃烧过程中形成NOx造成环境问题,而且在许多工业过程中还会导致催化剂的中毒。脱氮对许多不同的精炼工艺非常重要,其中加氢脱氮(HDN)是炼油工业除去油品馏分中含氮化合物的重要催化过程。加氢处理技术也是获得清洁油品的关键1。杂环类含氮化合物HDN反应网络中通常会出现苯胺类化合物,通过它们进一步发生HDN反应从而最终脱除氮。苯胺类中的C―N键断裂受到苯环空间位阻效应以及共轭效应的影响,因此选择合适催化剂提高苯胺类化合物加氢脱氮效率,是石油工业行业以及化工研究者面临的难题。
August 19,2015;Revised:November 16,2015;Published on Web:November 24,2015.
O641
*Corresponding author.Email:luxq@upc.edu.cn;Tel:+86-532-86983372.
The project was supported by the National Natural Science Foundation of China(21303266)and Petro China Innovation Foundation,China (2013D-5006-0406).
国家自然科学基金(21303266)和中国石油科技创新基金(2013D-5006-0406)资助项目
©Editorial office ofActa Physico-Chimica Sinica
猜你喜欢
能源化工(2022年1期)2023-01-14
高等学校化学学报(2022年4期)2022-06-10
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
西安文理学院学报(自然科学版)(2016年4期)2016-12-19
中国烟草学报(2016年3期)2016-11-23
质谱学报(2015年5期)2015-03-01
