
十二烷基苯磺酸钠和无机阳离子之间相互作用的密度泛函理论研究
2016-11-08 06:00刘志宏范成成张田田纪贤晶陈生辉孙霜青胡松青中国石化胜利油田有限公司地质科学研究院山东东营5705中国石油大学华东理学院山东青岛66580山东省高校新能源物理与材料科学重点实验室山东青岛66580
物理化学学报 2016年2期
刘志宏 范成成 张田田 纪贤晶 陈生辉 孙霜青, 胡松青,,*(中国石化胜利油田有限公司地质科学研究院,山东东营5705;中国石油大学(华东)理学院,山东青岛66580;山东省高校新能源物理与材料科学重点实验室,山东青岛66580)
十二烷基苯磺酸钠和无机阳离子之间相互作用的密度泛函理论研究
刘志宏1范成成2张田田2纪贤晶2陈生辉2孙霜青2,3胡松青2,3,*
(1中国石化胜利油田有限公司地质科学研究院,山东东营257015;2中国石油大学(华东)理学院,山东青岛266580;3山东省高校新能源物理与材料科学重点实验室,山东青岛266580)
研究阴离子表面活性剂和阳离子之间的相互作用对于理解阴离子表面活性剂的沉淀和溶解现象具有十分重要的理论和实际意义,但关于两者相互作用的相关理论模型鲜有报道。本文采用密度泛函理论(DFT)方法研究了十二烷基苯磺酸根阴离子(DBS-)与阳离子(Na+,Mg2+和Ca2+)在溶液内及气/液界面处的相互作用。在溶液内,在两种不同溶液环境中(水相和正十二烷)构建DBS-/阳离子相互作用模型,并对其进行优化。结果表明,DBS-能够与阳离子以双齿结构稳定结合。DBS-与阳离子的结合能不仅取决于参与的无机盐离子种类,还与溶剂的性质有关。在气/液界面处,DBS-与六个水分子相互作用形成的水合物DBS-∙6H2O最为稳定。但是,无机盐离子的引入会严重破坏DBS-∙6H2O水合物的水化层结构。本文定义无量纲参量def用来对水化层结构的变化程度进行评价。无机盐离子对DBS-∙6H2O水化层结构破坏程度的顺序为:Ca2+>Mg2+> Na+。电荷分析结果表明水化层在十二烷基苯磺酸钠(SDBS)头基与阳离子的相互作用中起了重要作用。
十二烷基苯磺酸钠;阳离子;相互作用;气/液界面;电荷分布;密度泛函理论
doi:10.3866/PKU.WHXB201512013
1 Introduction
Anionic surfactants are the most common surfactant products, whichhavebeenextensivelyappliedinmanyfields,suchasfroth flotation,detergency,purification,soliddispersion,pharmaceutical, enhanced oil recovery(EOR),etc1-4.However,mineral cations, such as Na+,Mg2+,and Ca2+,can significantly affect the macroscopic properties of surfactants,including surface tension,solubility,aggregation morphology,etc5-7.It potentially causes the precipitation and even ineffectiveness of anionic surfactants in many applications8,9.Therefore,it is of great significance to investigating the interaction behaviors between anionic surfactants and cations.
Over the past several years,experimental studies10-12and molecular dynamics(MD)simulations13-16on the interactions between anionic surfactants and inorganic salts have been reported.Bordes et al.10studied the interactions of three dicarboxylic amino acidbased surfactants with calcium ions based on the surface tension measurement and the nuclear magnetic resonance(NMR)analysis. They inferred that dodecylaminomalonate and dodecylaspartate could form intramolecular chelates with calcium ions,while dodecylglutamate preferred to form an intermolecular complex. Pereira et al.12studied the interactions of calcium ions with carboxylate anions under different alkyl chain lengths through turbidity,conductivity,and potentiometric measurements.They concluded that the electrostatic effect plays an important role in ordering the carboxylate anions before precipitation.MD simulations mainly emphasize the effect of cations on the self-assembly of surfactants at the interface or in the solution,supplying a detailed and atomistic insight into the three dimensional structures of the studied systems.However,both experimental techniques and MD simulations are incapable of accurately describing the structural and electrostatic properties.
Quantum mechanics(QM)technique has been employed into a variety of studies of surfactants17-21,but seldom reported on the study of the interactions of surfactants with ions.Zieliński and Szymusiak22performed density functional theory(DFT)calculations on the stability of water clusters surrounding the headgroup of alkyl-ammonium surfactant in the presence of different counterions.They found that stable water clusters could form if no less than three water molecules were contained.Yuan et al.23,24investigated the effect of cation types on the structure and charge distributions of both the anionic and zwitterionic surfactants using DFT method and conductor-like screening model(COSMO). Vlachy et al.25studied the interactions of the charged surfactant headgroups with aqueous ions based on computational studies with a combined method of molecular dynamics simulation and an ab initio calculation.They proposed a Hofmeister-like ordering of charged headgroups to describe and qualitatively predict ionheadgroup interaction for a wide range of systems and applications.
Upon dissolving in water,partial surfactant molecules are capable of adsorbing at the air/water interface,while the rest can present in the solution in the form of monomer or micelle,as shown in Fig.1.Unlike the surfactant monomer in the solution,the hydrophilic headgroups of the surfactant molecules at the air/water interface tend to adsorb on the interface while the hydrophobic tails are excluded from the water into the air.Additionally,the water molecules around the surfactant headgroup are thought to play an important role in the interactions between the surfactants and the inorganic salts according to MD simulations13,14.It is hence necessary to investigate the interactions of the surfactants and the cations both inside the solution and at the air/water interface.For the surfactant molecules in the micelle,the hydrophilic headgroups are in direct contact with water molecules while the rest are located in a water-free environment.It is reasonable to consider the surfactant molecule in the micelle as the case of that adsorbed at the air/water interface.
DFT is an extensively used method in investigating the hydrogen systems26-28and the structural transformation caused by the participation of ions29.In this paper,DFT method was used to investigate the molecular properties of the surfactant sodium dodecylbenzenesulfonate(SDBS),as well as the interactions between the dodecylbenzenesulfonate anion(DBS-)and different cations(Na+,Mg2+,and Ca2+).The binding mechanism and binding strength were comparatively studied inside the water solution and the n-dodecane solution separately in order to obtain the influence of the solvents.At the air/water interface,a hydrated complex of DBS-was constructed to describe the adsorbed surfactant molecule at the air/water interface.The effect of the cations on the structure and charge distribution of the hydrated complex was discussed as well. Mg2+>Na+.Acharge analysis reveals that the hydration shell plays an important role in the interactions between the sodium dodecyl benzene sulfonate(SDBS)headgroup and the cation.
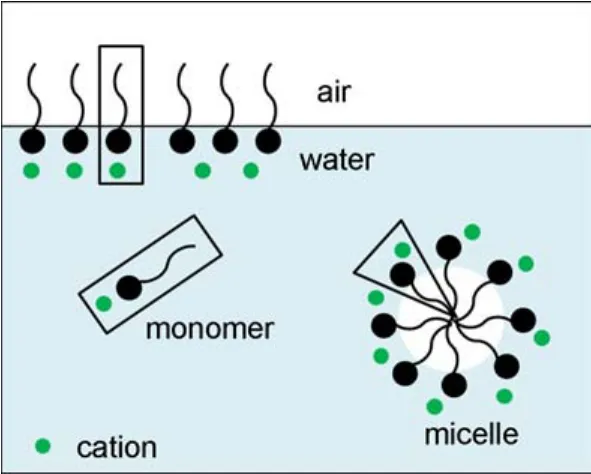
Fig.1 Schematic diagram of the surfactant/cation system in the aqueous solution with mineral cations
2 Simulation method and technical details
In this work,all DFT calculations were carried out using the generalized gradient approximation(GGA)with the Perdew-Burke-Ernzerhof(PBE)functional.The all-electron Kohn-Sham wave functions and double numerical plus polarization(DNP)30,31numerical basis set were selected.To minimize errors,the energies mentioned were all corrected with zero point vibrational energy (ZPVE corrected)22.Atomic charges were calculated using the Mulliken population analysis.All calculations were performed using Material Studio software(Accelrys,Inc).
In the process of the crude oil displacement,surfactants distribute in both water and oil phases.As the main constituent of crude oil,n-dodecane can be used to represent the oil phase in laboratory experiments or theoretical simulations.Therefore, water and n-dodecane environments were selected in this work and described by COSMO which is a well-established continuum solvation model incorporating solvation effects into the quantum mechanical calculation32.The dielectric constant of the water in the COSMO model was 78.54,while the dielectric constant of the ndodecane was set to 2.02.
In the construction of hydrated complexes of DBS-with different numbers of water molecules,the monohydrated complex was first determined by comparing the energy of all monohydrated configurations and choosing the one with the lowest energy.For the dihydrated complex,a second water molecule was added to the chosen monohydrated complex and then the dihydrated configuration with the lowest energy was found.By repeating this procedure,the hydrated complexes of DBS-with different numbers of water molecules(n=1-8)were constructed,as shown in Fig.2.
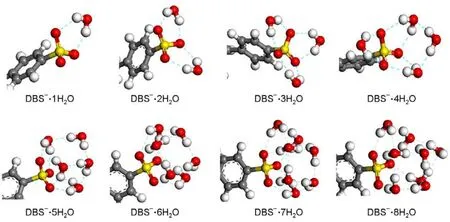
Fig.2 Configurations of the DBS-∙nH2O complexes(n=1-8)
The second order difference of interaction energy is usually used to estimate the stability of clusters33,34.The first and second order difference of the interaction energy between the DBS-and the water molecules(i.e.Δ1D(n)and Δ2D(n),respectively)could be calculated according to the following equations:
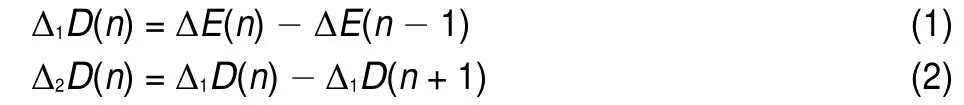
where ΔE(n)is the interaction energy between DBS-and n water molecules.Δ1D(n)indicates the ability to form the DBS-∙nH2O complex.Alarger Δ1D(n)means that it is easier to form the DBS-∙nH2O complex.Similarly,Δ1D(n+1)indicates the ability to form the DBS-∙(n+1)H2O complex.Δ2D(n)is the difference between Δ1D(n)and Δ1D(n+1),and it can be used to evaluate the stability of the DBS-∙nH2O complex.
3 Results and discussion
3.1Molecular properties of the single surfactant molecule
The fully optimized geometry of DBS-is shown in Fig.3.It can be seen that the DBS-exhibits a non-planar geometry.The angle between the long hydrocarbon chain and the line connecting the sulfur atom(S15)and the alpha-carbon(C7)is 144.3°.The length of the hydrocarbon chain(C7…H51)is 1.5009 nm.Table 1 lists the geometry parameters of the sulfonate group.The bond lengths of S15―O16,S15―O17,and S15―O18 are 0.1486,0.1488,and 0.1488 nm,respectively.In addition,the angle of∠O―S―O ranges from 112.7°to 113.3°.Considering the approximate bond length of S―O and angle of O―S―O,the sulfonate group is supposed to be a delocalized structure.The S―O bonds have the same chemical nature,which can also be supported by the results of bond order calculated using Mayer population analysis35.
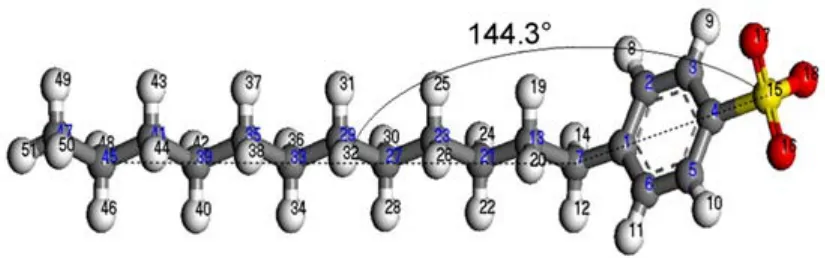
Fig.3 Geometry of the fully optimized DBS-
According to the frontier molecular orbital theory36,the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO)can be used to determine the way that the molecule interacts with other species.Fig.4 shows the electronic density distributions of HOMO and LUMO for DBS-.The HOMO is located on the sulfonate group with the energy of-0.20754 a.u.,while the LUMO mainly distributes over the sixmembered aromatic phenyl group with the energy of-0.05298 a.u.This indicates that the headgroup is highly active.

Table1 Geometry parameters of the optimized DBS-
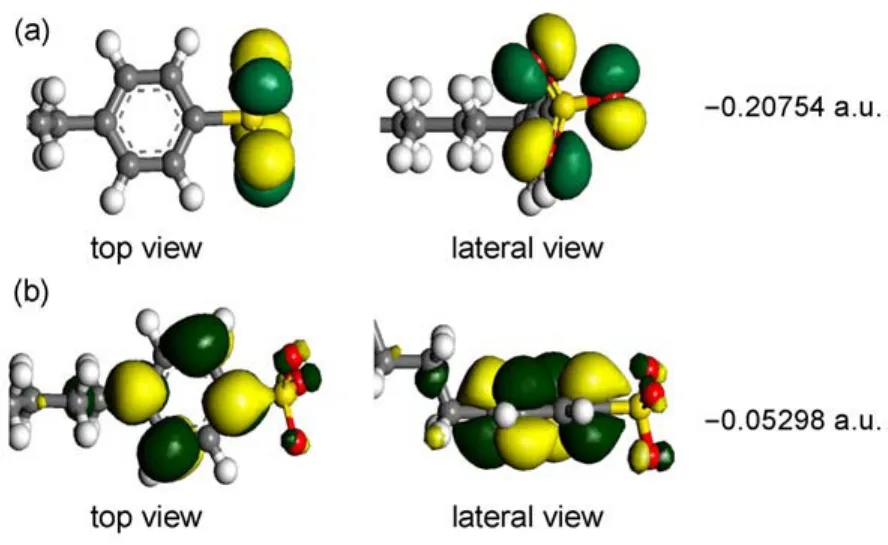
Fig.4 Isosurfaces of the frontier orbitals
The contributions of individual atoms to the frontier orbital are shown in Fig.5.The oxygen atoms in the sulfonate group make a contribution of nearly one hundred percent to the HOMO,and the contributions of O16,O17,and O18 are 33.5%,33.2%,and 32.9%,respectively.Therefore,these oxygen atoms in the headgroup will act as the reaction sites,and donate electrons when the DBS-interacts with the cations,water molecules or positively charged solid surfaces.It should be pointed out that,the difference of the oxygen atoms in the sulfonate group will not be considered. In addition to the carbon atoms in the phenyl group,atoms in the sulfonate group,and hydrogen atoms in the α-methylene group also contribute to the LUMO,which agrees well with the results displayed in the isosurfaces.
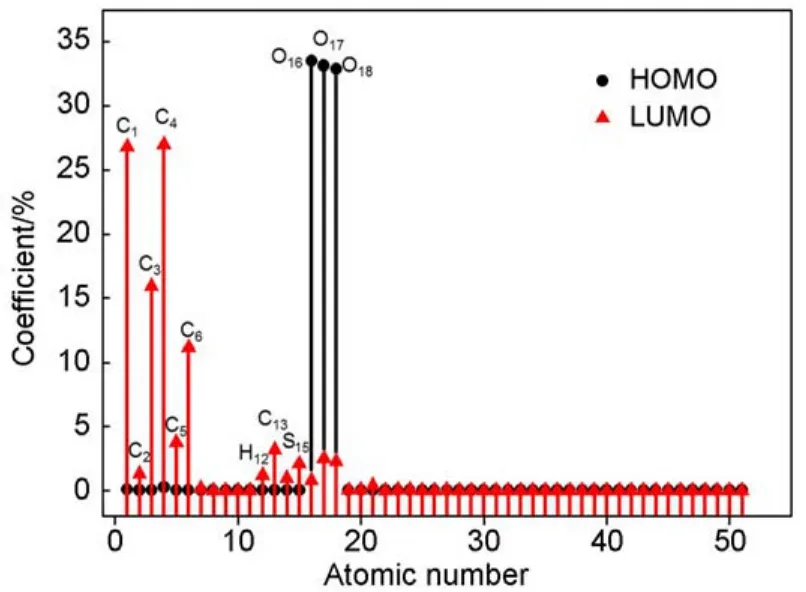
Fig.5 Atomic contributions of individual atoms to the frontier orbitals of DBS-
3.2Surfactant-cation interaction inside the solution
Based on the structural characteristics of the DBS-,three models were built to investigate the interaction between the DBS-and the cation inside the solution.The interaction models were defined by different atomic ratios of the oxygen atom to the cation (i.e.monodentate,bidentate,and tridentate structures).Every initial model was determined by changing the distances between the oxygen atoms and the cation and choosing the model with the lowest energy.
3.2.1Water solution
The initial and optimized DBS-/Mg2+systems inside the water solution are shown in Fig.6.Obviously,all the interaction models fall into a similar configuration with the ratio of the oxygen atom binding stably with the cation in a bidentate form.Mg2+is located in the plane composed of the sulfur atom and two oxygen atoms, and the distances between Mg2+and the two oxygen atoms are approximately equal.It indicates that Mg2+tends to bind with DBS-in a bidentate form inside the water solution.This might be due to the fact that DBS-would form a stable four-membered ring with Mg2+.Similar interactions of DBS-with Na+and Ca2+were also studied.
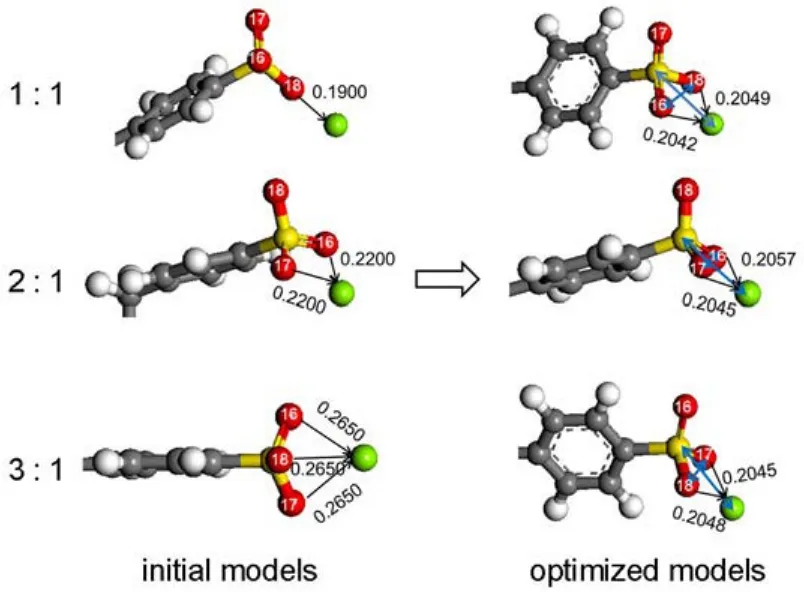
Fig.6 Initial and optimized models of DBS-/Mg2+inside the water solution
Table 2 lists the related geometry parameters and binding energies of DBS-/cation interaction models inside the water solution. It can be found that Na+and Ca2+exhibit a same binding form with DBS-as Mg2+.The binding energy ΔE,which represents the energy change before and after the interaction,is calculated by the following formula:

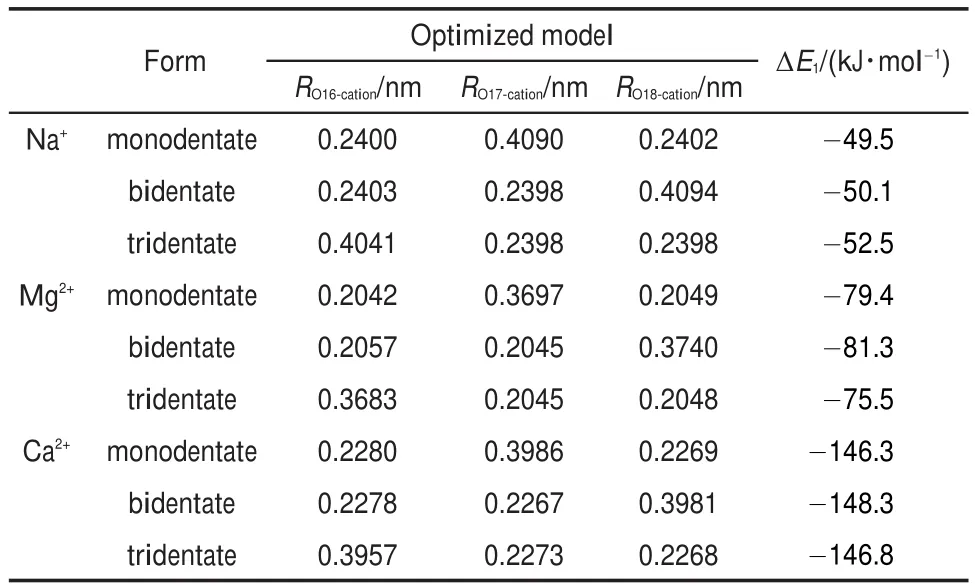
Table2 Geometry parameters and binding energies(ΔE1)of the DBS-/cation interaction models inside the water solution
where Esaa/cationis the total energy of the optimized model,Esaaand Ecationare the energies of the surfactant and the cation,respectively. The interaction is spontaneously,thus ΔE must be negative in value.The greater the absolute value of ΔE is,the more stable thebinding configuration is.
Inside the water solution,the binding energies of DBS-/Na+, DBS-/Mg2+,and DBS-/Ca2+are approximately-50.7,-78.7,and-147.1 kJ∙mol-1,respectively.It can be seen that the absolute values of ΔE1(DBS-/Ca2+)and ΔE1(DBS-/Mg2+)are larger than ΔE1(DBS-/Na+).Once the calcium soap or magnesium soap of SDBS is formed inside the solution,they would be more difficult to be dissolved compared with the sodium soap.This can give a tentative explain to the precipitation of anionic surfactants in the condition of high salinity.

Table3 Geometry parameters and binding energies(ΔE2)of the DBS-/cation interaction models inside the n-dodecane solution
The interactions between anionic surfactants and cations are primarily dominated by the electrostatic effect and the hydration effect.Ca2+and Mg2+have one more positive charge than Na+,thus they bind more strongly with DBS-.For identically charged Mg2+and Ca2+,Mg2+with a smaller ionic radius37and a higher electron density is supposed to have greater electrostatic effect.However, the absolute value of ΔE1(DBS-/Ca2+)is greater than ΔE1(DBS-/ Mg2+)with a difference of 68.4 kJ∙mol-1.In the aqueous solution with strong polarity,Mg2+was verified to have a strong hydration effect which can weaken the electrostatic effect.In this work, COSMO describes the response of a solvent medium by generating screening charge on the cavity surface(or solvent-accessible surface)of the solutes.Inside the water solution,Mg2+can induce more screening charge than Ca2+,which hence decreases the electrostatic effect more greatly.
3.2.2n-Dodecane solution
The geometry parameters and binding energies of the DBS-/ cation systems inside the n-dodecane solution are listed in Table 3.All the optimized interaction models are in the bidentate form as those inside the water solution.However,the absolute values of binding energies are significantly greater.For example,the binding energy of DBS-/Mg2+inside the n-dodecane solution reaches up to approximately-780 kJ∙mol-1.This is due to the low dielectric constant of the n-dodecane solution.Inside the ndodecane solution,the hydration effect is too weak and there is not enough induced screening charge to weaken the electrostatic effect.In addition,the ΔE2(DBS-/Mg2+)is more negative than ΔE2(DBS-/Ca2+)and ΔE2(DBS-/Na+).In contrary to the result inside the water solution,Mg2+binds more strongly with the DBS-than Ca2+.As mentioned before,the hydration effect inside the ndodecane solution is weak and the electrostatic effect plays a crucial role in the interaction.The electrostatic effect between Mg2+and DBS-is much stronger than that between Ca2+and DBS-, which leads to the difference.
3.3Surfactant -cation interaction at the air/water interface
3.3.1The most stable hydrated complex of DBS
In Section 2,the hydrated complexes of DBS-with different numbers of water molecules were built.Fig.7 shows the relationship between the second order difference of the interaction energy Δ2D(n)and the number of water molecules(n).It can be seen that the absolute value of Δ2D(6)is the highest,which indicates that the DBS-∙6H2O is the most stable model.In other words,SDBS tends to interact with six water molecules at the air/ water interface.The water molecules surround the hydrophilic headgroup and provide a good representation for the hydration shell.
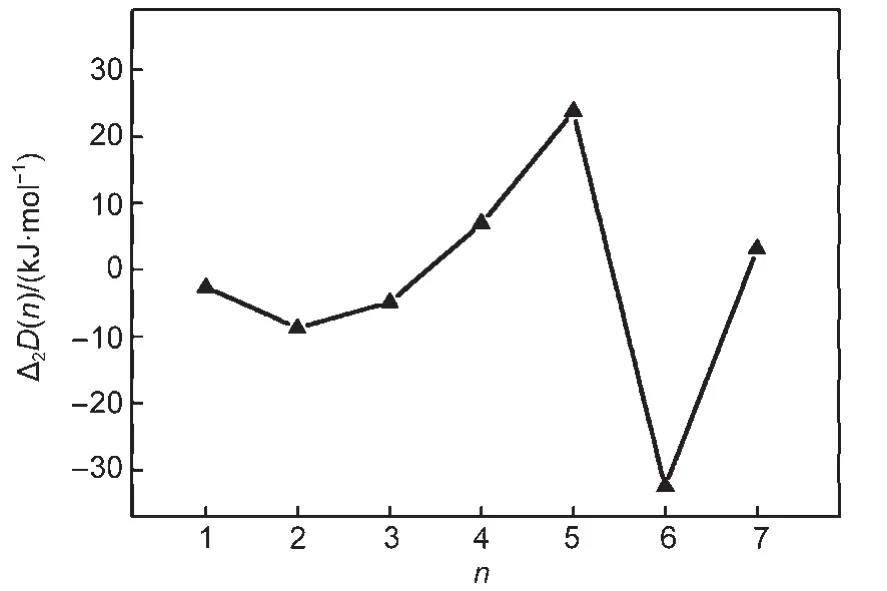
Fig.7 Relationship between the second order difference of interaction energy(Δ2D(n))and the number of water molecules(n)
The structure of the DBS-∙6H2O complex is shown in Fig.8. W1,W2,W3,and W6 form hydrogen bonds with the headgroup directly,while W4 and W5 interact with the headgroup through the bridging of the other water molecules.The geometry parameters of formed H-bonds are listed in Table 4.The Y―X distance and H…Xdistance are in the range of 0.2765-0.3118 nmand 0.1768-0.2434 nm,respectively(where X is a hydrogen donor,and Y is the atom accepting the hydrogen).The Y―H…X angle ranges from 130.7°to 171.8°.It is well known that the energy of hydrogen bond can be qualitatively estimated according to the H…X length38.The average length of H…X between the headgroup and water molecules is 0.2051 nm,longer than those between water molecules(0.1845 nm).Therefore,H-bond between the polar headgroup and water molecule is weaker than those between water molecules.
3.3.2Effect of the cation on the structure of the DBS-∙6H2O complex
To reduce the influence of the water molecules on the results, we constructed ideal interaction models in which all cations are located between the oxygen atoms(O16 and O18)in the headgroup.The optimized results with Na+,Mg2+,and Ca2+are pre-sented in Fig.9.Overall,Na+affects the adjacent water molecules (W1,W2,and W4)apparently,while Mg2+and Ca2+disrupt the original configuration of the whole hydration shell.
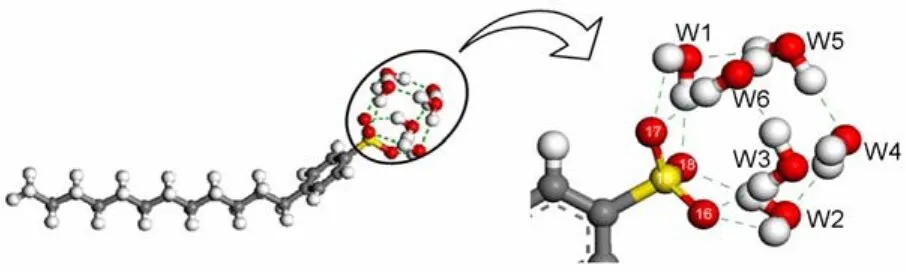
Fig.8 Arrangement of the water molecules in the DBS-∙6H2O complex
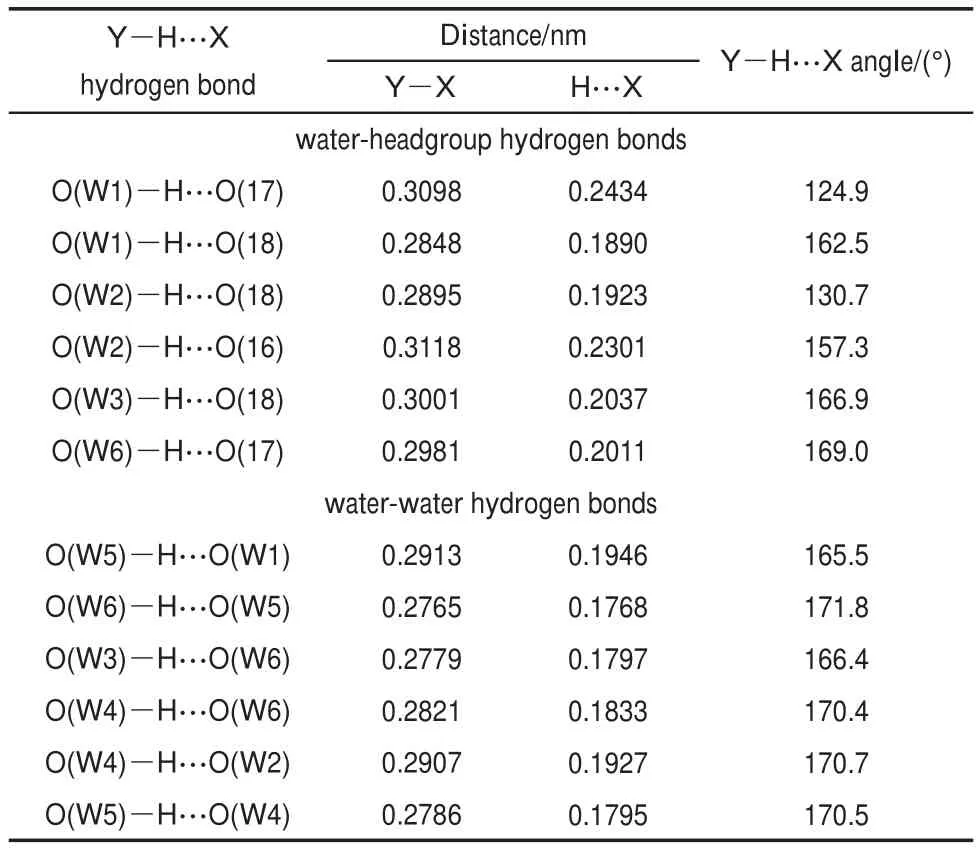
Table4 Geometry parameters of H-bonds in the DBS-∙6H2O complex
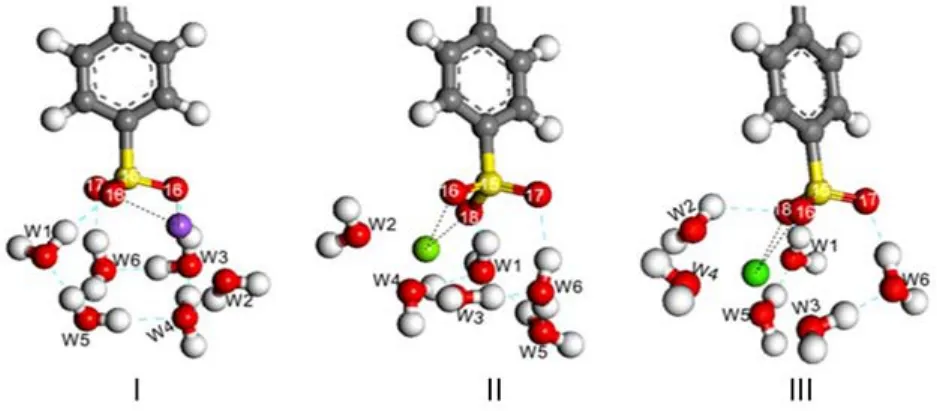
Fig.9 Optimized interaction models
Table 5 lists the distances between the cation and oxygen atoms (O16 and O18).The RO16-cationand RO18-cationare close to each other, suggesting that the cations still bind with the headgroup in the bidentate form as that found inside the solution.The little difference between RO16-cationand RO18-cationmight be resulted from the influence of the adjacent water molecules.Moreover,the oxygen atoms in the water molecules are found to be close to the cation, while the hydrogen atoms have relative far distances from the cation.This distribution would be favorable to the formation of H-bonds with water molecules in the bulk phase.

Table5 RO16-cation,RO18-cation,and structural deformation parameter (def)of the hydration shell in the optimized interaction models
def,a dimensionless parameter,was proposed to quantitatively describe the structural deformation of the hydration shell.
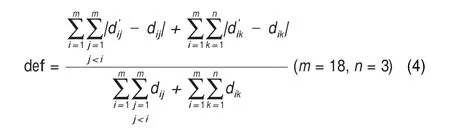
where,dijandare the distances between atom i and atom j in the hydration shell before and after the introduction of a cation, respectively.Similarly,dikandare the distances between atom i in the hydration shell and oxygen atom k in the headgroup before and after the introduction of a cation,respectively.It includes two parts,(1)the total variation of distances between different atoms in the hydration shelland(2)the total variation of distances between every atom in the hydration shell and every oxygen atom in the headgroupThe results of def(listed in Table 5)clearly reveal that the disturbing degree of the each cation to the hydration shell can be arranged in a decreasing order as follows:Ca2+>Mg2+>Na+.It suggests that the divalent cation can influence the hydration shell more greatly than the monovalent cation due to the stronger electrostatic interactions.Moreover,Ca2+with larger ionic radius has a more apparent effect on the hydration shell than Mg2+.
3.3.3Effect of the cation on the charge distribution of the DBS-∙6H2O complex
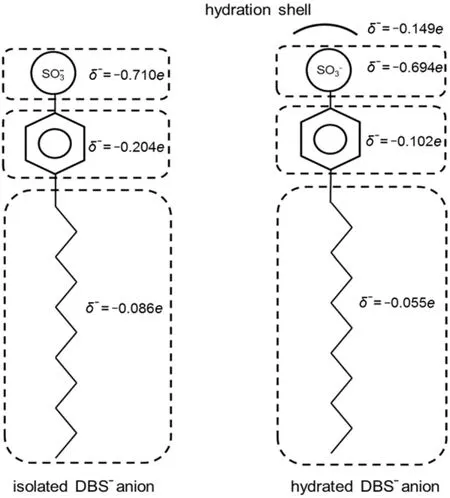
Fig.10 Charge distributions of the isolated DBS-and the hydrated DBS-∙6H2O complex
For the ionic surfactant systems,the charge distribution was often mentioned to investigate the electrostatic effect or charge related physico-chemical properties39,40.For the sake of brevity,the DBS-∙6H2O complex was described in different sub-molecular groups,i.e.the hydration shell(-0.149e),the sulfonic group (-0.694e),the aromatic phenyl group(-0.102e),and the alkyl tail (-0.055e)(Fig.10).Compared with the isolated DBS-,the negative charge of corresponding sub-molecular groups in the DBS-∙6H2O complex is reduced.About 14.9%negative charge transfers from the isolated surfactant molecule to the hydration shell.It should be more accurate to estimate the charge distribution of the ionic surfactant with consideration of the impact of water molecules.
Fig.11 shows the charge redistribution of the DBS-∙6H2O complex after introducing different cations.In general,Mg2+causes the most notable decrease in the total charge of the hydrated complex(-1e to-0.414e),then Ca2+(-1e to-0.525e)and Na+(-1e to-0.826e).The negative charge in hydrophobic parts (i.e.the alkyl tail and the aromatic phenyl group)decreases in the value and even turns to positive.Interestingly,the negative charge in the headgroup increases after introducing the cation.This is because the cation with positive charge attracts the negative charge of the surfactant anion concentrating in the headgroup.But the hydration shell around the headgroup plays an important role in preventing the charge transfer between the surfactant anion and cation.That is why the hydration shell is more positively charged.
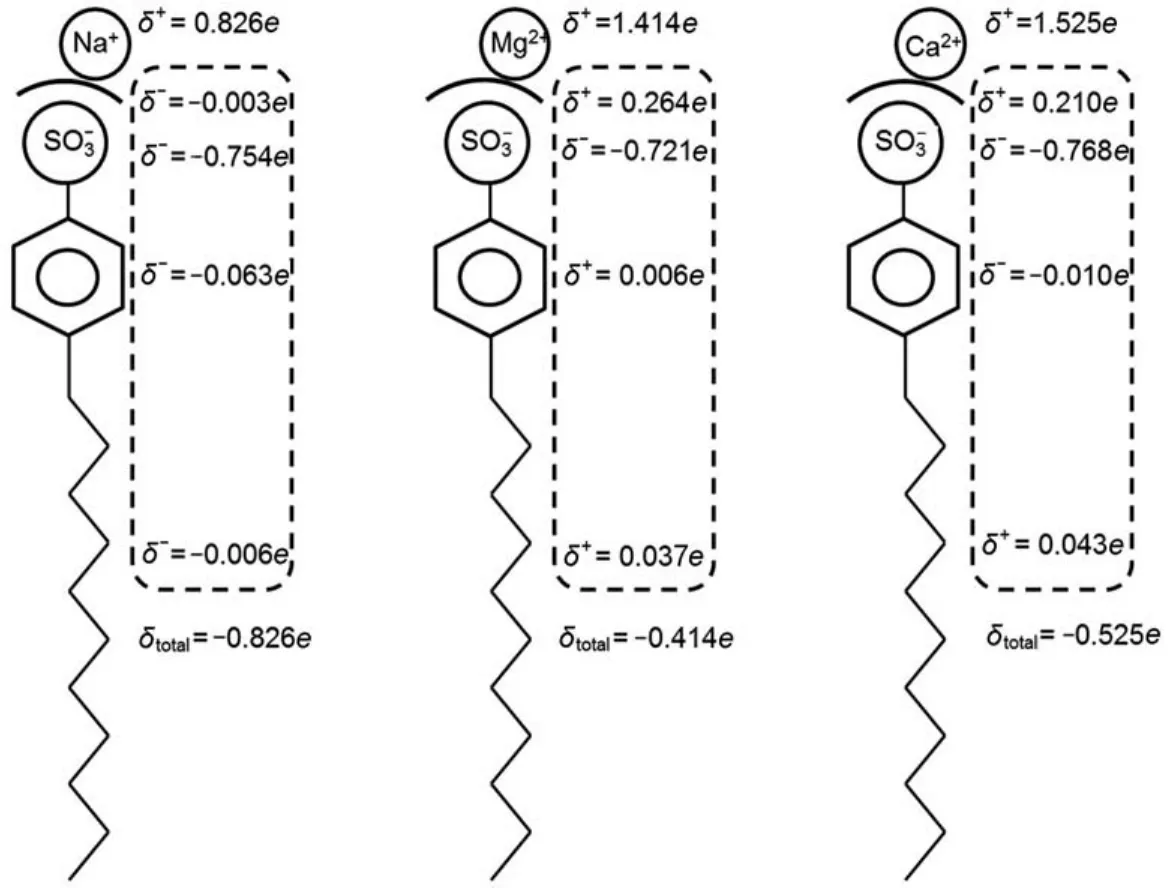
Fig.11 Charge redistributions of the DBS-∙6H2O complex after introducing different cations
4 Conclusions
In this work,two different models have been built to investigate the interactions of the surfactant SDBS and the mineral cations both inside the solution and at the air/water interface.Firstly,the structural analysis of single DBS-anion indicates that the sulfonate headgroup is highly active and different oxygen atoms in the DBS-have the same interaction with the cation.Inside the solution,cations tend to bind with DBS-in a bidentate form,and the binding energy of the DBS-and the cation depends on the characteristics of both the participating cation and the solvent medium.The binding energy,which implies the binding strength of the surfactant and the cation,can be explored to correlate with the solubility of formed metal salts or estimate the salt tolerance of different surfactants.Although this method is at the price of a continuum treatment of the solvent,it still can be generalized in order to provide some microscopic information.At the air/water interface,the hydrated complex DBS-∙6H2O is determined to represent the adsorbed surfactant molecule.The water molecules around the hydrophilic headgroup provide a good representation for the hydration shell.This can be taken as a wise way to represent the complicated task of the adsorption at the air/water interface in theory.A dimensionless parameter,def,is proposed to evaluate the deformation extent of the hydration shell caused by the participation of cation,and the disturbing degree of the cation follows the order:Ca2+>Mg2+>Na+.Furthermore,the hydration shell is shown to play an important role in the interaction between the surfactant headgroup and the cation based on the results of the charge analysis.
References
(1)Rosen,M.J.;Kunjappu,J.T.Surfactants and Interfacial Phenomena;Wiley:New Jersey,2012.
(2)Hirasaki,G.;Miller,C.;Puerto,M.SPE Journal 2011,16,889. doi:10.2118/115386-PA
(3)Cserháti,T.;Forgács,E.;Oros,G.Environment International 2002,28,337.doi:10.1016/S0160-4120(02)00032-6
(4)Gomez,V.;Ferreres,L.;Pocurull,E.;Borrull,F.Talanta 2011, 84,859.doi:10.1016/j.talanta.2011.02.009
(5)Yu,D.;Wang,Y.;Zhang,J.;Tian,M.;Han,Y.;Wang,Y. Journal of Colloid and Interface Science 2012,381,83.doi: 10.1016/j.jcis.2012.05.016
(6)Santos,F.K.G.;Neto,E.L.B.;Moura,M.C.P.A.;Dantas,T. N.C.;Neto,A.A.D.Colloids and Surfaces A: Physicochemical and Engineering Aspects 2009,333,156.doi: 10.1016/j.colsurfa.2008.09.040
(7)Vakarelski,I.U.;Dushkin,C.D.Colloids and Surfaces A: Physicochemical and Engineering Aspects 2000,163,177.doi: 10.1016/S0927-7757(99)00306-4
(8)Stellner,K.L.;Scamehorn,J.F.Langmuir 1989,5,70.doi: 10.1021/la00085a014
(9)Noïk,C.;Bavière,M.;Defives,D.Journal of Colloid and Interface Science 1987,115,36.doi:10.1016/0021-9797(87) 90006-3
(10)Bordes,R.;Tropsch,J.;Holmberg,K.Journal of Colloid and Interface Science 2009,338,529.doi:10.1016/j. jcis.2009.06.032
(11)Bordes,R.;Holmberg,K.Colloids and Surfaces A: Physicochemical and Engineering Aspects 2011,391,32.doi: 10.1016/j.colsurfa.2011.03.023
(12)Pereira,R.F.P.;Valente,A.J.M.;Fernandes,M.;Burrows,H. D.Physical Chemistry Chemical Physics 2012,14,7517.
(13)Zhao,T.T.;Xu,G.Y.;Yuan,S.L.;Chen,Y.;Yan,H.The Journal of Physical Chemistry B 2010,114,5025.
(14)Yan,H.;Guo,X.L.;Yuan,S.L.;Liu,C.B.Langmuir 2011, 27,5762.doi:10.1021/la1049869
(15)Sammalkorpi,M.;Karttunen,M.;Haataja,M.The Journal of Physical Chemistry B 2009,113,5863.doi:10.1021/jp901228v
(16)Domínguez,H.Langmuir 2009,25,9006.doi:10.1021/ la900714a
(17)Yan,P.;Xiao,J.X.Colloids and Surfaces A:Physicochemical and Engineering Aspects 2004,244,39.doi:10.1016/j. colsurfa.2004.06.023
(18)Motamedi,M.;Bathaie,S.Z.;Hemmateenejad,B.;Adjloo,D. Journal of Molecular Structure:Theochem 2004,678,163.
(19)Kocherbitov,V.;Veryazov,V.;Söderman,O.Theochem 2007, 808,111.doi:10.1016/j.theochem.2006.12.043
(20)Aime,C.;Plet,B.;Manet,S.;Schmitter,J.M.;Huc,I.;Oda, R.;Sauers,R.R.;Romsted,L.S.The Journal of Physical Chemistry B 2008,112,14435.doi:10.1021/jp802801r
(21)Zhu,M.;Ge,F.;Zhu,R.;Wang,X.;Zheng,X.Chemosphere 2010,80,46.doi:10.1016/j.chemosphere.2010.03.044
(22)Zieliński,R.;Szymusiak,H.International Journal of Quantum Chemistry 2004,99,724.
(23)Li,Z.Q.;Yan,H.;Song,X.W.;Yuan,S.L.;Pan,B.L.;Wang, L.J.Acta Chimica Sinica 2011,69,898.[李振泉,延辉,宋新旺,苑世领,潘斌林,王丽娟.物理化学学报,2011,69, 898.]doi:10.3866/PKU.WHXB20110431
(24)Cao,X.L.;Lü,K.;Cui,X.H.;Shi,J.;Yuan,S.L.Acta Physico-Chimica Sinica 2010,26,1959.[曹绪龙,吕凯,崔晓红,石静,苑世领.物理化学学报,2010,26,1959.]doi: 10.3866/PKU.WHXB20100706
(25)Vlachy,N.;Jagoda-Cwiklik,B.;Vácha,R.;Touraud,D.; Jungwirth,P.;Kunz,W.Advances in Colloid and Interface Science 2009,146,42.doi:10.1016/j.cis.2008.09.010
(26)Iype,E.;Nedea,S.V.;Rindt,C.C.M.;Steenhoven,A.A.V.; Zondag,H.A.;Jansen,A.P.J.The Journal of Physical Chemistry C 2012,116,18584.doi:10.1021/jp3025649
(27)Fumino,K.;Peppel,T.;Geppert-Rybczynska,M.;Zaitsau,D. H.;Lehmann,J.K.;Verevkin,S.P.;Kockerling,M.;Ludwig, R.Physical Chemistry Chemical Physics 2011,13,14064.doi: 10.1039/c1cp20732f
(28)Thanthiriwatte,K.S.;Hohenstein,E.G.;Burns,L.A.;Sherrill, C.D.Journal of Chemical Theory and Computation 2010,7, 88.
(29)Shishkin,M.;Ziegler,T.The Journal of Physical Chemistry C 2009,113,21667.doi:10.1021/jp905615c
(30)Inada,Y.;Orita,H.Journal of Computational Chemistry 2008, 29(2),225.
(31)Delley,B.The Journal of Chemical Physics 1990,92(1),508. doi:10.1063/1.458452
(32)Klamt,A.;Schuurmann,G.Journal of the Chemical Society, Perkin Transactions 1993,2,799.
(33)Li,X.J.;Su,K.H.Theor.Chem.Acc.2009,124,345.doi: 10.1007/s00214-009-0618-9
(34)Bandyopadhyay,D.;Sen,P.The Journal of Physical Chemistry A 2010,114,1835.doi:10.1021/jp905561n
(35)Mayer,I.Journal of Quantum Chemistry 1986,29,477.
(36)Gece,G.Corrosion Science 2008,50,2981.doi:10.1016/j. corsci.2008.08.043
(37)Kiriukhin,M.Y.;Collins,K.D.Biophysical Chemistry 2002, 99,155.doi:10.1016/S0301-4622(02)00153-9
(38)Shishkin,O.V.;Gorb,L.;Leszczynski,J.The Journal of Physical Chemistry B 2000,104,5357.doi:10.1021/jp993144c
(39)Zhao,G.X.;Zhu,B.Y.;Dou,Z.P.;Yan,P.;Xiao,J.X. Colloids and Surfaces A:Physicochemical and Engineering Aspects 2008,327,122.doi:10.1016/j.colsurfa.2008.06.014
(40)Huibers,P.D.T.Langmuir 1999,15,7546.doi:10.1021/ la990367l
Density Functional Theory Study of the Interaction between Sodium Dodecylbenzenesulfonate and Mineral Cations
LIU Zhi-Hong1FAN Cheng-Cheng2ZHANG Tian-Tian2JI Xian-Jing2CHEN Sheng-Hui2SUN Shuang-Qing2,3HU Song-Qing2,3,*
(1GeologicalScientificResearchInstitute,ShengliOilfieldCompany Ltd.,Dongying 257015,Shandong Province,P.R.China;2CollegeofScience,ChinaUniversityofPetroleum(EastChina),Qingdao266580,ShandongProvince,P.R.China;3KeyLaboratory ofNewEnergyPhysics&MaterialsScienceinUniversitiesofShandong,Qingdao266580,ShandongProvince,P.R.China)
Investigating the interactions between anionic surfactants and cations is of great theoretical and practical significance to understanding the precipitation and solubility of anionic surfactant products but relevant theoretical interaction models are seldom reported.In this paper,the density functional theory(DFT)method was used to investigate the interactions of the dodecylbenzenesulfonate anion(DBS-)with Na+,Mg2+,and Ca2+both in the solution and at the air/water interface.In the solution,DBS-/cation interaction models were built and optimized with consideration of two different solutions(i.e.water and n-dodecane).The results indicate that DBS-can bind stably with the cations in a bidentate form.The binding energy of the DBS-/cation depends on the properties of both the participating cation and the solvent.At the air/water interface,DBS-formed a stable hydrated complex with six water molecules(i.e.DBS-∙6H2O).However,the structure of DBS-∙6H2O was greatly disturbed by the introduction of the cation.A dimensionless parameter,def,was proposed to evaluate the deformation extent of the hydration shell.The degree of disturbance by the cations follows the order:Ca2+>
July 28,2015;Revised:November 27,2015;Published on Web:December 1,2015.
SDBS;Cation;Interaction;Air/water interface;Charge distribution; Density functional theory
O641
*Corresponding author.Email:ccupc@upc.edu.cn;Tel:+86-532-86983170.
The project was supported by the PetroChina Innovation Foundation,China(2015D-5006-0213)and Fundamental Research Funds for the Central Universities,China(14CX02221A,14CX06157A).
中国石油科技创新基金(2015D-5006-0213)及中央高校基本科研业务费专项资金(14CX02221A,14CX06157A)资助项目
©Editorial office ofActa Physico-Chimica Sinica
猜你喜欢
无机盐工业(2021年12期)2021-12-15
化工设计(2020年2期)2020-05-01
中国洗涤用品工业(2019年9期)2019-09-25
广西植物(2016年5期)2016-06-17
纺织导报(2015年10期)2016-01-04
西安石油大学学报(自然科学版)(2015年4期)2015-12-16
疑难病杂志(2014年12期)2014-04-16
中国洗涤用品工业(2012年8期)2012-03-20
中国洗涤用品工业(2012年8期)2012-03-20
中国洗涤用品工业(2012年8期)2012-03-20
