
线状及涡纹状色素过多沉积症一例
2016-11-06 09:09杨彪尚元元喻楠焦亚宁王立鹏董灵娣扈容英王媛葛新红
中华皮肤科杂志 2016年2期
杨彪 尚元元 喻楠 焦亚宁 王立鹏 董灵娣 扈容英 王媛 葛新红
750000银川,宁夏医科大学(杨彪);宁夏医科大学总医院皮肤科(尚元元、喻楠、焦亚宁、王立鹏、董灵娣、扈容英、王媛、葛新红)
线状及涡纹状色素过多沉积症一例
杨彪 尚元元 喻楠 焦亚宁 王立鹏 董灵娣 扈容英 王媛 葛新红
750000银川,宁夏医科大学(杨彪);宁夏医科大学总医院皮肤科(尚元元、喻楠、焦亚宁、王立鹏、董灵娣、扈容英、王媛、葛新红)
患者女,18岁,主诉全身出现色素沉着斑18年,间断腹痛7年,患者出生1周后面颈部、躯干、四肢逐渐出现黑褐色斑,色斑随年龄增长逐渐增多、扩大,呈线状、条索状或漩涡状,色素斑发生前无红斑、水疱或瘙痒。间断腹痛7年,偶有恶心、呕吐,无腹泻。既往无癫痫发作史,否认放射性物质、毒物接触史,月经初潮年龄17岁,月经期2 d,月经间期2~3个月,量少。父母均体健,非近亲结婚,母亲正常怀孕、分娩,怀孕期间无服药史,否认家族中类似病例。
体检:形体消瘦、中度营养不良,神清、语利,智力发育正常,牙齿排列整齐,无锥形齿。双眼瞳孔等大、正圆,对光反射灵敏,视力正常,视野无缺损。余系统检查未见明显异常。皮肤科检查:面、颈部可见线状或片状褐色色素沉着斑,躯干、四肢见沿Blaschko线分布的线状、条索状、漩涡状、片状或不规则形褐色色素沉着斑,见图1。黏膜、毛发及指趾甲未见异常。背部褐斑组织病理检查示基底细胞色素增加,部分基底细胞有空泡化改变,真皮浅层可见噬色素细胞(图2)。实验室检查:尿常规及肝肾功能未见异常。血常规提示轻度贫血,骨髓活检、左手关节X线片、甲状腺功能全套均未见明显异常。电子结肠镜检查示距肛门60 cm处可见一较大不规则5 cm×4 cm息肉致肠镜无法通过,未见结肠及直肠黏膜其他异常,组织病理:(结肠)黏膜组织显示慢性炎症。见图3。结合临床诊断:线状及涡纹状色素过多沉积症。
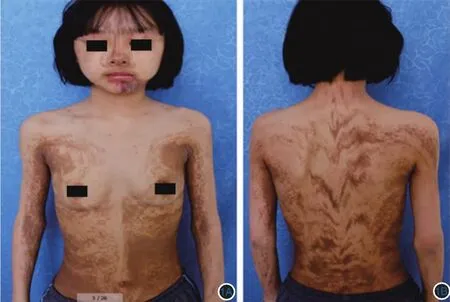
图1 患者临床表现 面部(1A)可见线状或片状淡褐色色素沉着斑,躯干、四肢(1B)可见沿Blaschko线分布的线状、漩涡状或泼墨状褐色色素沉着斑

图2 背部褐斑组织病理 表皮基底细胞色素增多,部分基底细胞空泡化改变,真皮上层可见少量噬色素细胞(HE×400)

图3 电子结肠镜检查 距肛门60 cm处可见一较大不规则5 cm×4 cm息肉,致肠镜无法通过。未见结肠及直肠黏膜其他异常
讨论 线状及涡纹状色素过多沉积症为胚胎发育时体细胞镶嵌所致,男孩和女孩均可见到,患者多于出生后1年内出现漩涡形和条纹状棕色色素沉着,病理主要表现为基底层黑素细胞增多,真皮内噬色素细胞极少,无色素失禁[1]。该患者除了皮肤上有色素沉着斑外,尚未发现有眼睛、牙齿、神经系统等病变,但表现为形体消瘦(体重36 kg),伴有肠息肉、间断腹痛。因为是单发肠息肉,也不能排除是偶然伴发。该患者背部褐色色素沉着斑呈“V”形且沿Blaschko线分布,临床上应与持久性色素失禁症相鉴别。
持久性色素失禁症是色素失禁症的异型,多见于女性[2]:自幼发生于躯干、四肢的黑褐色斑点,随着年龄及身体的增长而增多扩大呈条纹状或漩涡状,至成年期褐色斑疹亦不消退,在出现褐色斑前皮肤无红斑、水疱及疣状增生史[3];该病罕见,检索维普科技期刊全文数据库(1989—2015)、万方数据 库 (1998—2015)、 中 国 知 网(1976—2015)、PubMed(1950—2015)显示国内有1例报道[2],国外未见报道。色素失禁症也称Bloch-Sulzberger综合征,是一种少见的单基因遗传性皮肤病[4],目前认为,该病是由NEMO/IKK-γ基因发生突变所致[5]。表现为出生后不久或出生时即有红斑、水疱,继之疣状炎症增生,褐色斑是继发性的,好发于躯干、上臂及大腿,历时数年,消退后不留痕迹[6]。上述皮损可同时发生亦可交替出现,病变亦可引起眼睛、牙齿、神经系统等病变[7-9]。临床上女性发病多见,男性多宫内死亡而少见[4]。
[1]刘玉梅,高歆婧,田歆,等.线状和漩涡状痣样过度黑素沉着病伴局限性掌跖角化病1例[J].中国皮肤性病学杂志,2014,28(8):826-828.Liu YM,Gao XJ,Tian X,et al.A case of linnear and whorled nevoid hypermelanosis complicated by focal palmoplantar keratoderma[J].Chin J Derm Venereol,2014,28(8):826-828.
[2]唐亚娟.异型色素失禁症1例报告[J].临床皮肤科杂志,1994,3:166.Tang YJ.Atypical form of incontinentia pigmenti:a case report[J].J Clin Dermatol,1994,3:166.
[3]曹元华,何秀珍,夏隆庆.Blaschko线的研究进展及其相关皮肤病[J].中华皮肤科杂志,2004,37(2):126-128.Cao YH,He XZ,Xia LQ.Advances in Blaschko′s lines and related dermatoses[J].Chin J Dermatol,2004,37(2):126-128.
[4]Hadj-Rabia S,Froidevaux D,Bodak N,et al.Clinical study of 40 cases of incontinentia pigmenti[J].Arch Dermatol,2003,139(9):1163-1170.DOI:10.1001/archderm.139.9.1163.
[5]Fusco F,Bardaro T,Fimiani G,et al.Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation[J].Hum Mol Genet,2004,13 (16):1763-1773.DOI:10.1093/hmg/ddh192.
[6]郝丽红,荣丽英,麻庆荣,等.新生儿色素失禁症临床特点分析[J].临床荟萃,2010,25(19):1711-1712,1652.Hao LH,Rong LY,Ma QR,et al.Clinical analysis of neonatals with incontinentia pigmenti[J].Clinical Focus,2010,25(19):1711-1712,1652.
[7]唐兰芳,邹朝春.色素失禁症临床分析[J].中华皮肤科杂志,2001,34(6):442.Tang LF,Zou CC.Clinical analysis of incontinentia pigmenti[J].Chin J Dermatol,2001,34(6):442.
[8]Goldberg MF.Macular vasculopathy and its evolution in incontinentia pigmenti[J].Ophthalmic Genet,1998,19(3):141-148.DOI:10.1076/opge.19.3.141.2190.
[9]Buinauskiene J,Buinauskaite E,Valiukeviciene S.Incontinentia pigmenti(Bloch-Sulzberger syndrome)in neonates[J].Medicina(Kaunas),2005,41(6):496-499.
2015-03-30)
(本文编辑:尚淑贤)
葛新红,Email:gexinhong.0101@163.com
10.3760/cma.j.issn.0412-4030.2016.02.017
猜你喜欢
山东冶金(2022年2期)2022-08-08
北方人(2020年22期)2020-12-02
疯狂英语·新悦读(2020年4期)2020-06-18
阅读(快乐英语中年级)(2019年11期)2019-09-10
汽车观察(2018年10期)2018-11-06
中外文摘(2017年19期)2017-10-10
中国卫生(2016年4期)2016-11-12
建材发展导向(2016年3期)2016-05-23
山东青年(2016年2期)2016-02-28
中国卫生(2014年4期)2014-12-06
