
超高效合相色谱-质谱法分析研究单甘酯乳化剂中单甘酯的主要组成
2016-11-01 07:11:09林春花刘德永范乃立夏剑辉廖维林江西师范大学国家单糖化学合成工程技术研究中心化学化工学院南昌33007
分析化学 2016年2期
林春花刘德永范乃立夏剑辉廖维林(江西师范大学国家单糖化学合成工程技术研究中心,化学化工学院,南昌 33007)
超高效合相色谱-质谱法分析研究单甘酯乳化剂中单甘酯的主要组成
林春花1刘德永1范乃立1夏剑辉*2廖维林1
(江西师范大学国家单糖化学合成工程技术研究中心1,化学化工学院2,南昌 330027)
建立了超高效合相色谱-质谱(UPC2-MS)快速分析3种单甘酯乳化剂(单油酸甘油酯、单亚油酸甘油酯、单硬脂酸甘油酯)中单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯和单亚油酸甘油酯等4种主要的单甘酯的方法,并比较了这3种不同类别的乳化剂中此4种单甘酯的含量差异。采用正己烷/异丙醇(7∶3,V/V)直接溶解样品,以ACQUITY UPC2BEH 2-EP色谱柱(100 mm×2.1 mm,1.7μm)为分析柱,以超临界CO2-甲醇/乙腈(1∶1,V/V)为流动相进行梯度洗脱,流速为1.0 mL/min。在电喷雾正离子模式下进行分析,外标法定量。结果表明:单棕榈酸甘油酯、单硬脂酸甘油酯、单亚油酸甘油酯在0.20~50 mg/L范围内具有良好线性,单油酸甘油酯在0.25~62.5 mg/L范围内具有良好的线性(相关系数不小于0.9983);定量限(S/N≥10)为0.018~0.046 mg/L;在3个加标水平下,样品的回收率在88.0%~110.5%,相对标准偏差为1.1% ~4.1%。本方法简单、快速、分离效果好,无需对单甘酯样品进行衍生化,为乳化剂中单甘酯的含量分析提供了一种新的色谱技术手段。
超高效合相色谱-质谱法;单甘酯;乳化剂;未衍生化
1 引言
乳化剂是一种能改善乳化体中各种构成相之间的表面张力,稳定乳浊液的添加剂。单甘酯是食品行业重要的乳化剂[1],按照主要组成脂肪酸(C16~C18)的名称可以将其分为单棕榈酸甘油酯、单硬脂酸甘油酯等饱和脂肪酸单甘酯和单油酸甘油酯、单亚油酸甘油酯等不饱和脂肪酸单甘酯[2,3]。一直以来,单甘酯的生产对象主要为饱和脂肪酸单甘酯,且单硬脂酸甘油酯使用的最多,它几乎成了单甘酯的代名词。近年来,研究发现不饱和脂肪酸单甘酯,具有更低的表面张力和更好的乳化能力[4,5],是一种更为优良的食品乳化剂和保健食品添加剂。因此,对单甘酯乳化剂制品中单甘酯的主要组成进行分析,将有助于产品质量的提高以及食品安全风险的控制。
单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯和单亚油酸甘油酯等单甘酯结构类似,分子量相差不大,沸点较高。现有的分析方法有化学滴定法[6]、TLC[7]、GPC[8]、GC[9~12]、HPLC-UV/RID/ ELSD[4,13~16]等。化学滴定法易使单甘酯的测定结果偏高,且不适合对样品中存在不饱和脂肪酸单甘酯进行分析[15,17];TLC操作简单,但定量分析不太准确[18,19];GPC只能测定单甘酯总量,不能对上述4种单甘酯进行完全分离;GC是较常用的方法,直接分析样品时需要高温测试条件,色谱柱的使用寿命大大降低,而间接分析样品时需要进行衍生化处理,过程繁琐且影响产物的准确测定[20];HPLC-ELSD法虽无需样品衍生化,但分析时间较长,检测限较高。近年来,随着UPLC色谱技术的发展,在一定程度上弥补了HPLC的不足。因此,建立一种更为简便且分离度更高的测定单甘酯的方法很有必要。
超高效合相色谱(UPC2)是基于超临界流体色谱(SFC)的基本原理而发展起来的一种新型色谱技术,它集合了SFC和UPLC两者的优势,开辟了分离科学的新领域[21]。分析现有研究成果发现,SFC是分析脂类物质方法中选择性较好的方法[22]。而UPC2继承和发展了SFC的优势,是新一代快速、准确分析单甘酯的技术手段。本研究就是利用UPC2对单甘酯乳化剂中4种主要的单甘酯进行分析测定,不但验证了UPC2方法的科学性和可行性,同时也为评价单甘酯乳化剂的品质提供了数据参考。
2 实验部分
2.1 仪器与试剂
ACQUITY UPC2系统(美国Waters公司),配Waters SQD 2质谱检测器。ACQUITY UPC2HSS C18SB色谱柱(100 mm×2.1 mm,1.7μm)、ACQUITY UPC2BEH色谱柱(100 mm×3 mm,1.7μm)、ACQUITY UPC2BEH 2-EP色谱柱(100 mm×2.1 mm,1.8μm)和ACQUITY UPC2CSHTMFluoro-Phenyl Column色谱柱(100 mm×3 mm,1.7μm)(美国Waters公司)。METTLER TOLEDO AL204型电子天平(瑞士METTLER公司)。PURELAB Ultra MK 2型超纯水仪(英国ELGA公司)。可调式移液器(上海大龙公司)。
4种单脂肪酸甘油酯标准品:单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯和单亚油酸甘油酯(纯度大于99%,Sigma Aldrich公司);甲醇、乙腈、正己烷和异丙醇(色谱纯,德国Merck公司);甲酸、甲酸铵(色谱级,上海安谱公司);醋酸铵(色谱级,美国Fluka公司);实验用水为超纯水。单硬脂酸甘油酯乳化剂、单油酸甘油酯乳化剂和单亚油酸甘油酯乳化剂均为市售。
2.2 实验条件
2.2.1 色谱条件色谱柱:ACQUITY UPC2BEH 2-EP;流动相:超临界CO2(A)和甲醇-乙腈(1∶1,V/V)(B)。洗脱梯度:0~2 min,3.5%~5%B;2~3 min,5%B;3~5 min,5%~12%B;5~6min,12%~3.5%B。柱温:45℃;样品室温度:10℃;背压:1.103×107Pa;流速:1.0 mL/min;进样体积:1 μL。
2.2.2 质谱条件离子源:ESI+;毛细管电压:3.0 kV;锥孔电压:30V;源温度:120℃;脱溶剂气温度:500℃;脱溶剂气体流速:800 L/h;锥孔气体流速:30 L/h;补偿溶剂:10 mmol/L甲酸铵溶液,流速为0.2 mL/min;选择离子监测模式下,单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯和单亚油酸甘油酯的m/z分别为348.50,376.56,374.54,372.56([M+NH4]+),提取离子色谱图如图1所示。

图1 4种单脂肪酸甘油酯标准品在ESI+模式下的提取离子色谱图Fig.1 Extracted ion chromatograms of the four monoglyceride standards in ESI+mode
2.2.3 混合标准溶液的配制分别准确称取单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯和单亚油酸甘油酯标准品,用正己烷/异丙醇(7∶3,V/V)溶解,依次配成浓度为1.000 g/L,1.000 g/L,1.250 g/L,1.000 g/L的单一标准品储备液。移取不同体积的各单甘酯标准溶液混合配制成混合标准溶液,再稀释成系列标准工作溶液。
2.2.4 样品制备分别称取0.01 g(精确至0.0001 g)单硬脂酸甘油酯乳化剂、单油酸甘油酯乳化剂和单亚油酸甘油酯乳化剂等样品,用正己烷-异丙醇(7∶3,V/V)溶解并定容至100 mL,经有机膜过滤后待测。
3 结果与讨论
3.1 色谱柱的选择
UPC2系统可以使用传统正相和反相色谱柱的填料,为开发分离方法提供了多种选择性。本实验首先考察了4种不同填料的ACQUITY UPC2色谱柱对单甘酯分离的影响,它们分别为BEH,BEH 2-EP,HSS C18SB及CSHFluoro-Phenyl。由图2可见,除HSS C18SB柱外,其它色谱柱的出峰顺序相似。BEH柱和BEH 2-EP柱对单甘酯混合物的选择性最好,都能够实现4种单甘酯的有效分离。但BEH 2-EP柱的极性小于BEH柱,更适合分离极性较大的单甘酯混合物,具有较理想的分析时间和选择性,因此本实验选用BEH 2-EP色谱柱。

图2 不同色谱柱对4种单脂肪酸甘油酯分离效果的影响Fig.2 Effect of different chromatographic columns on the separation of four monoglycerides Peaks:1.1-Monopalmitin;2.1-Monostearin;3.1-Monoolein;4.1-Monolinolein.
3.2 洗脱梯度的选择
洗脱梯度的变化会对目标物的保留时间和分离度产生一定的影响。本实验考察了3组不同的洗脱梯度的方法(方法1:0~2 min,1.5%~5%B;2~3 min,5%~12%B;3~4 min,12%B;4~6min,12% ~1.5%B,方法2:0~2 min,3.5%~5%B;2~3 min,5%B;3~5 min,5%~12%B;5~6 min,12%~3.5%B,方法3:0~2 min,3.5%~12%B;2~3 min,12%B;3~6 min,12%~3.5%B)。由图3可见,共溶剂中流动相B的含量越高,保留时间越短(方法1>方法2>方法3);洗脱梯度的斜率越大,分离度越小(方法2>方法1>方法3)。综合考虑分离度和保留时间,最终选用方法2的洗脱梯度。
3.3 柱温的选择
改变柱温会在一定程度上影响UPC2的分离效果,所以实验中以混合标准品为对象,在改性剂为甲醇-乙腈(1∶1,V/V)、流速为1.0 mL/min和背压1.103×107Pa的条件下,考察柱温(35℃,45℃,55℃)对分离效果的影响。由图4可见,随着柱温升高,目标物的保留增大,这是因为升高温度会降低CO2的密度,从而减弱其洗脱能力,导致化合物的保留值增大;同时目标物的分离度随着温度的升高而有所增加,当升高至55℃时增幅不大,因此选用45℃柱温。

图3 不同的洗脱梯度对4种单甘酯分离度和保留时间的影响Fig.3 Effect of elution gradients on resolution and retention time of four monoglycerides
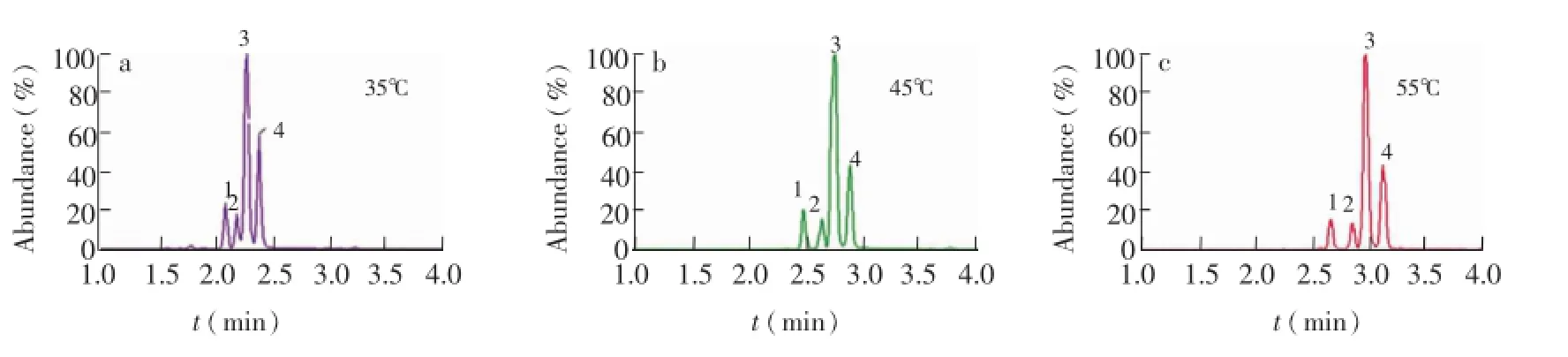
图4 温度对4种单干酯分离效果的影响Fig.4 Effect of temperature on the separation of four monoglycerides
3.4 背压的选择
背压的改变也会在一定程度上影响UPC2的分离效果,所以在改性剂为甲醇-乙腈(1∶1,V/V)、流速1.0 mL/min和柱温45℃的条件下,考察了不同的系统背压(1.103×107~1.378×107Pa)对分离效果的影响。结果如图5所示,背压升高造成CO2的密度和黏度增加,从而导致流动相的溶剂化能力和洗脱能力提高,进而使得化合物的保留时间缩短;同时,背压升高对分离度影响不大。因此,选择背压为1.103×107Pa时,目标物的分离效果与保留效果较好。
3.5 MS中补偿溶剂的选择
在LC-MS分离中,一般会使用补偿溶剂,以改善峰形和促进目标物的分子离子化。在相同的梯度洗脱条件下,流速为0.2 mL/min时,实验考察了分别含0.1%(V/V)甲酸、10 mmol/L甲酸铵和10 mmol/L醋酸铵的甲醇作为补偿溶剂时4种单甘酯的分离情况。由图6可见,使用甲酸铵、醋酸铵等缓冲盐作补偿溶剂,总离子流图(TIC)的响应值分别为1.40×108和1.33×108,明显高于使用甲酸的补偿溶剂。因此,最终选用甲醇-10 mmol/L甲酸铵作为补偿溶剂,目标物的离子化效率最好,响应灵敏度最高。

图5 背压对4种单干酯分离效果的影响F i g . 5 E f f e c t o f b a c k p r e s s u r e o n t h e s e p a r a t i o n o f f o u r m o n o g l y c e r i d e s
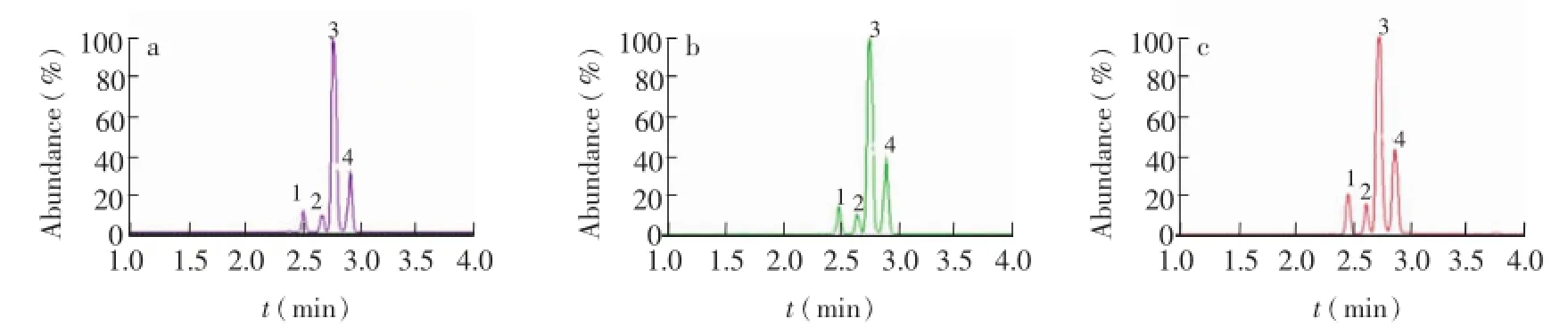
图6 补偿溶剂对4种单干酯分离效果的影响Fig.6 Effect of compensation solvent on the separation of the four monoglycerides
3.6 方法评价
配制不同质量浓度的混合标准溶液,在优化的色谱条件下进行检测,以峰面积(y)对质量浓度(x,mg/L)进行线性回归,得到线性回归方程。向已知含量的乳化剂样品中分别添加4种成分的单甘酯标准品,设置3个添加水平,每个加标水平重复测定6次,并计算加标回收率和精密度。按信噪比S/N≥10计算得到分析方法的定量限(LOQ)。结果(表1)表明,单棕榈酸甘油酯、单硬脂酸甘油酯、单亚油酸甘油酯在0.20~50.0 mg/L内线性关系良好,而单油酸甘油酯在0.25~62.5 mg/L内线性关系良好(R2≥0.9983),3个加标水平的回收率在88.0%~110.5%,RSD为1.1%~4.1%,LOQ为0.018~0.046 mg/L,可满足食品乳化剂中单甘脂的测定要求。

表1 4种单脂肪酸甘油酯的方法评价结果Table 1 Evaluation of the method for the 4 monoglycerides
运用本方法对市售的3种单甘酯乳化剂(单硬脂酸甘油酯乳化剂、单油酸甘油酯乳化剂、单亚油酸甘油酯乳化剂)样品进行检测(表2),结果表明,3种单甘酯乳化剂中4种1-单甘酯的含量和比例因类别不同而差异较大。其中,单油酸甘油酯乳化剂和单亚油酸甘油酯乳化剂都是以1-油酸单甘酯和1-亚油酸单甘酯等不饱和1-单甘酯为主,它们在上述2种乳化剂中的总含量分别为71.73%和85.92%。单硬脂酸甘油酯乳化剂是以1-单棕榈酸和1-单硬脂酸甘油酯等饱和1-单甘油酯为主,它们在该乳化剂中的总含量高达91.01%。3种样品的色谱图如图7所示。
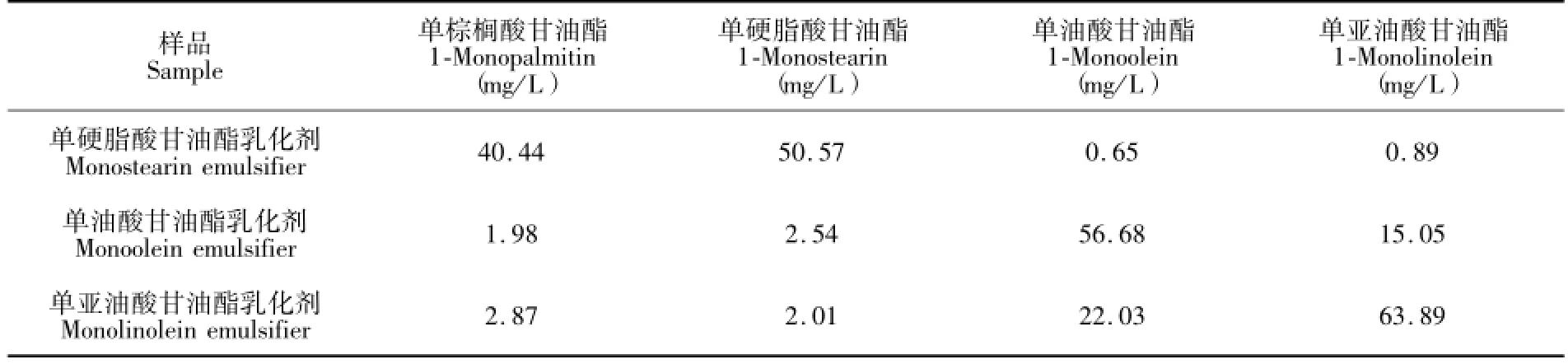
表2 实际样品的分析结果Table 2 Analytical results of real sample

图7 单硬脂酸甘油酯乳化剂(a)、单油酸甘油酯乳化剂(b)、单亚油酸甘油酯乳化剂(c)中4种单甘酯的色谱图Fig.7 UPC2chromatograms of 4 monoglycerides in the monostearin emulsifier(a),monoolein emulsifier(b)and monolinolein emulsifier(c)
4 结论
通过对色谱柱、洗脱梯度、柱温、背压、补偿溶剂等条件的优化和考察,建立了超高效合相色谱-质谱法快速分析单甘酯乳化剂中4种单甘酯的分析方法。本方法采用超临界CO2作为主要流动相,它具有与液体相近的密度、与气体接近的粘度及扩散系数等优良性能。同以往色谱分析方法相比,本方法获得了灵敏度更高和分析速度更快的结果,为快速、准确分析单甘酯的主要组成提供了新的技术手段,同时也为评价单甘酯乳化剂的品质提供了数据参考。
1 LI Yan,LIU Jun-Hai.Cereals&Oils,2011,(3):7-11
李燕,刘军海.粮食与油脂,2011(3):7-11
2 SHI Hong,GUO Hong.China Oils and Fats,2000,25(4):38-41
时宏,郭洪.中国油脂,2000,25(4):38-41
3 NI Yong-Quan.The Third Member of China Food Additive Association Congress and the Ninth China International Exhibition of Food Additives and Ingredients of Academic Papers,2005:3
倪永全.中国食品添加剂协会第三届会员代表大会暨第九届中国国际食品添加剂和配料展览会学术论文集,2005:3
2.1.2 料液比 一般情况下,在料液比较大时花青素提取效果较好。山竹果皮花青素提取量随料液比升高而降低,而溶液中花青素含量随料液比升高而升高。花青素提取量曲线与溶液中花青素含量曲线出现一个交点,此时,料液比为1∶28。当料液比过大时则会造成不必要的溶剂浪费和能源消耗,故正交试验料液比选用1∶25~1∶35。
4 LIU Yan-Feng,HUANG Hui-Hua.Modern Food Science and Technology,2012,28(1):86-90
刘艳丰,黄惠华.现代食品科技,2012,28(1):86-90
5 MOU Ying,CUI Hai-Ping,YANG Tian-Kui.China Food Additives,2012,S1:88-92
牟英,崔海萍,杨天奎.中国食品添加剂,2012,S1:88-92
6 YI Xia,FAN Tie.China Oils and Fats,2008,33(9):73-75
栾霞,樊铁.中国油脂,2008,33(9):73-75
7 MENG Xiang-He,ZHANG Yin-Jun,MAO Zhong-Gui.China Oils and Fats,2004,29(1):44-46
孟祥河,章银军,毛忠贵.中国油脂,2004,29(1):44-46
8 ZHANG Bo,SI Zhi-Kun.Shandong Chem Industry,2003,32(6):27-28
张博,司芝坤.山东化工,2003,32(6):27-28
9 FENG Feng-Qin,YANG Hong-Jie.Chinese Food Science,2005,5(1):62-66
冯凤琴,杨宏杰.中国食品学报,2005,5(1):62-66
10 WANG Peng,SUN Yang,LIU Zhe-Yi,FU Yu-Fei,PAN Zai-Fa,WANG Li-Li.Chinese J.Anal.Chem.,2011,39(9):1427-1431
王鹏,孙杨,刘哲益,傅宇飞,潘再法,王丽丽.分析化学,2011,39(9):1427-1431
11 WAGN Li-Li,WAGN Yong,HU Chang-Ying,ZHANG Yun.China Oils and Fats,2011,36(7):75-79
王丽丽,汪勇,胡长鹰,张云.中国油脂,2011,36(7):75-79
12 MA Yu-Song,JIA Hai-Tao,YAO Chun-Yi,GUO Chun-Hai.Journal of Instrumental Analysis,2013,32(8):1012-1015
马育松,贾海涛,姚春毅,郭春海.分析测试学报,2013,32(8):1012-1015
13 Takano S,Kondoh Y.J.Am.Oil Chem.Soc.,1987,64(7):1001-1003
14 Di Nicola G,Pacetti M,Polonara F,Santori G,Stryjekb R.J.Chromatogr.A,2008,1190(1):120-126
15 LIU Liu,LU Zong-Feng,LI Li-Jun,LAI Ying-Biao.Physical Testing and Chemical Analysis Part B:Chemical Analysis,2011,47(12):1468-1469
刘柳,陆宗峰,李利军,赖映标.理化检验(化学分册),2011,47(12):1468-1469
16 Fedosova S N,Fernandesb N A,Firdausa M Y.J.Chromatogr.A,2014,1326:56-62
17 YANG Jing,HU Dao-Hua.Science and Technology of Food Industry,2004,25(7):127-128
杨菁,胡道华.食品工业科技,2004,25(7):127-128
18 Freedman B,Pryde E H,Kwolek W F.J.Am.Oil Chem.Soc.,1984,61:1215-1220
19 Fagan P,Wijesundera C,Watkins P.J.Chromatogr.A,2004,1054(1):251-259
20 Csernica S N,Hsu J T.Energy Fuels,2010,24:6131-6141
21 LIN Chun-Hua,FAN Nai-Li,RUI Pei-Xin,XIA Jian-Hui,LIAO Wei-Lin,YANG Shao-Ming.Chinese J.Anal.Chem.,2015,43(1):75-80
林春花,范乃立,芮培欣,夏剑辉,廖维林,杨绍明.分析化学,2015,43(1):75-80
22 Bamba T,Lee J W,Matsubara A,Fukusaki E.J.Chromatogr.A,2012,1250:212-219
This work was supported by the National Science and Technology Support Project(Nos.2012BAE07B00,2014BAE13B02 )
Determination of Main Composition of Fatty Acid Monoglyceride in Monoglyceride Emulsifier Using Ultra Performance Convergence Chromatography-Mass Spectrometry
LIN Chun-Hua1,LIU De-Yong1,FAN Nai-Li1,XIA Jian-Hui*2,LIAO Wei-Lin1
(National Monosaccharide Chemical Synthesis Engineering Research Center1,Chemistry and Chemical Engineering Department2,Jiangxi Normal University,Nanchang 330027,China)
A fast analytical method was developed for the determination of main composition of fatty acid monoglycerides,including monopalmitin,monostearin,monoolein and monolinolein,in monoglycerede emulsifiers by ultra-performance convergence chromatography-massspectrometry. Theircontentswere compared in the 3 emulsifiers from different sources.The sample was directly dissolved with n-hexane/isopropanol(7∶3,/V)The chromatographic separation was performed on the ACQUITY UPC2BEH 2-EP column(2.1 mm×100 mm,1.7μm)using the mobile phases of carbon dioxide and methanol/acetonitrile (1∶1,V/V)solution with gradient elution.The separated compounds were detected by MS detector in positive electrospray ionization(ESI+)and quantified by external standard method.The results showed that the calibration curves of monopalmitin,monostearin,monoolein and monolinolein were linear in the range of 0.20-50 mg/L,0.20-50 mg/L,0.25-62.5 mg/L and 0.20-50 mg/L,respectively,with correlation coefficients not less than 0.9983.The limits of quantification(S/N≥10)of the four fatty acid monoglycerides were 0.018-0.046 mg/L.The average recoveries for the four monoglycerides at three spiked levels were 88.0%-110.5%with relative standard deviations of 1.1%-4.1%.The proposed method showed high performance,good selectivity and fast analysis for the underivatized monoglyceride samples.It would provide a new chromatographic technology for the content analysis of monoglycerides in the emulsifier.
Ultra-performance convergence chromatography-mass spectrometry;Fatty acid monoglyceride;Emulsifier;Underivatization
14 July 2015;accepted 21 October 2015)
10.11895/j.issn.0253-3820.150558
2015-07-14收稿;2015-10-21接受
本文系国家科技支撑计划项目(Nos.2012BAE07B00,2014BAE13B02)资助
*E-mail:xjh2168@126.com
猜你喜欢
核农学报(2020年11期)2020-12-04 06:53:42
中国蜂业(2019年3期)2019-04-03 11:02:16
西南国防医药(2016年6期)2016-12-01 06:01:14
中国塑料(2016年2期)2016-06-15 20:30:00
当代化工研究(2016年7期)2016-03-20 16:21:54
池州学院学报(2015年3期)2016-01-05 01:13:12
肉类研究(2014年2期)2014-04-29 00:44:03
食品工业科技(2014年15期)2014-03-11 18:17:45
湖南农业科学(2014年3期)2014-02-27 14:28:21
食品科学(2013年8期)2013-03-11 18:21:24
