
超高效液相色谱-串联质谱法测定生活饮用水中10种农药残留
2016-10-25 08:16朱建丰陈军缪英
化学分析计量 2016年5期
朱建丰,陈军,缪英
(江阴市疾病预防控制中心,江苏江阴 214400)
超高效液相色谱-串联质谱法测定生活饮用水中10种农药残留
朱建丰,陈军,缪英
(江阴市疾病预防控制中心,江苏江阴 214400)
采用超高效液相色谱-串联质谱法测定生活饮用水中10种农药残留。水样直接进样,选用Waters ACQUITY UPLC®BEH C18柱分离,以乙腈-0.1%甲酸溶液为流动相进行梯度洗脱,质谱选用多反应监测模式分析,10种农药的质量浓度在0.5~50 μg/L 范围内与色谱峰面积呈线性相关,检出限(3S/N)在0.03~0.50 μg/L之间,测定值的相对标准偏差在1.14%~9.91%之间,加标回收率在91.6%~107.1%之间。该法简便准确,适用于生活饮用水中农药残留的检测。
超高效液相色谱—串联质谱法;生活饮用水;农药残留
莠去津、毒死蜱、乐果、敌敌畏、灭草松、呋喃丹、对硫磷、甲基对硫磷、马拉硫磷和2,4-滴是《生活饮用水卫生标准》(GB 5749-2006)要求检测的项目指标[1]。这10种目标物在GB 5750.9-2006 《生活饮用水标准检验方法》中规定的检测方法分别为(1)莠去津,水样经二氯甲烷萃取、浓缩、挥干和定容之后,用高效液相色谱紫外检测器(HPLC-UV)检测;(2)毒死蜱、乐果、敌敌畏、对硫磷、甲基对硫磷和马拉硫磷,水样经二氯甲烷萃取、浓缩后,用气相色谱火焰光度检测器(GC-FPD)检测;(3)灭草松和2,4-滴,水在酸性条件下经乙酸乙酯萃取,然后在碱性条件下用碘甲烷溶液酯化,生成较易挥发的甲基化衍生物,用毛细管柱气相色谱-电子捕获器(GC-ECD)分别测定;(4)呋喃丹,水样过滤后注入HPLC,经柱后衍生反应,用荧光检测器(FLR)测定。目前,国内实验室普遍参考GB 5750.9-2006检测水中的10种目标物。水样都需要进行萃取、浓缩或者衍生化处理,然后再用气相色谱法或液相色谱法[2]进行检测,检测步骤繁琐,回收率低,耗时长[3]。国内相关研究主要集中于水中有机氯、有机磷[4]、三嗪类[5]等单一农药监测及调查方面,近年来,农药多残留检测主要采用气相色谱-质谱[6-8]和液相色谱-质谱[9-17]联用技术,这些方法大多需提前对水样进行提取和净化处理。运用超高效液相色谱-串联质谱技术,样品无需预处理,直接测定水样中莠去津等10种农药残留的相关报道较少。
笔者建立了利用超高效液相色谱-串联质谱法测定生活饮用水莠去津等10种农药残留的检测方法,该法灵敏度高,直接进样测定,大幅提高了工作效率,检测结果更为快速准确。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱仪:Waters I-Class型,美国Waters 公司;
三重四级杆质谱仪:Waters TQ-S型,配有电喷雾离子源(ESI)和Masslynx数据处理系统,美国Waters 公司;
乐果,敌敌畏,呋喃丹,灭草松,莠去津,2,4-滴,甲基对硫磷,马拉硫磷,对硫磷,毒死蜱标准储备溶液:质量浓度均为100 mg/L,以甲醇为溶剂,农业部环境保护科研检测所;
混合标准溶液:1 000 μg/L,吸取10种标准储备液各100 μL,均加入到预先装有甲醇的10 mL A级容量瓶中,用甲醇定容至标线,于4℃冰箱内保存,临用前用乙腈-水(3∶7)溶液稀释至所需质量浓度;
甲酸:纯度≥98.0%,德国默克公司;
乙腈、甲醇:色谱纯,纯度≥99.9%,德国默克公司;
微孔滤膜:0.22 μm有机相滤膜;
实验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
色谱柱:Waters ACQUITY UPLC®BEH C柱
18(2.1 mm×50 mm,1.7μm);柱温:10℃;进样体积:10 μL;流动相:A为乙腈,B为0.1%甲酸水溶液;梯度洗程序见表1。
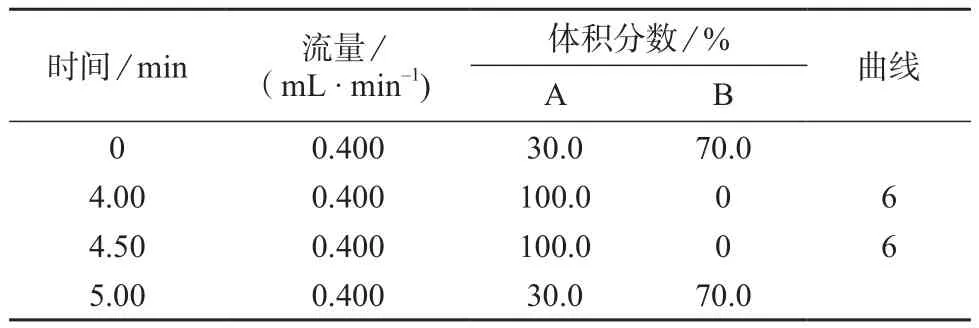
表1 梯度洗脱条件
1.2.2 质谱条件
电喷雾离子源(ESI);采集方式:多反应监测(MRM);毛细管电压:3.50 kV;离子源温度:150℃;雾化温度:500℃;雾化气流量:700 L/h;二级碰撞气:氩气,流量为0.15 mL/min;其它质谱参数见表2。
1.3 实验方法
直接取约2 mL水样,通过0.22 μm微孔滤膜导入进样瓶中上机测定,在1.2仪器工作条件下测定。
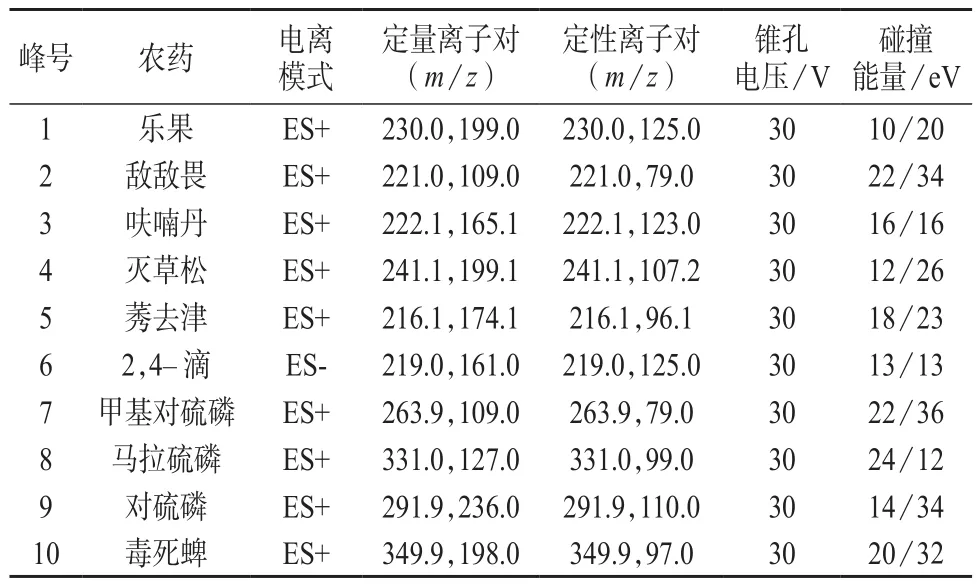
表2 质谱参数
2 结果与讨论
2.1 色谱条件优化
首先对Waters ACQUITY UPLC®RBEH系列C18柱、C8柱、Phenyl柱和Shield RP18柱进行了比较,其中C18柱对10种目标物的分离和保留效果最好,且大部分目标物都获得了最高响应值,因此实验选择C18柱。
分别选择乙腈和甲醇作为流动相A,对10种目标物的混合溶液洗脱情况进行比较,发现用乙腈时洗脱速度快,分离情况良好,其响应值大于甲醇作为流动相时的响应值,因此选择乙腈为流动相。
采用正离子电离模式进行质谱检测时,为提高离子化效率,使目标物容易质子化而带正电荷,需在流动相中加入一定量的甲酸。分别选择含0,0.01%,0.05%,0.1%,0.5%甲酸水溶液为流动相。试验结果显示,随着甲酸浓度增加,目标物的响应值也增加,但随着甲酸浓度的进一步增加,目标物的响应趋于稳定。考虑分离度、峰形和响应强度,选择0.1%甲酸水溶液为流动相。
选择初始流动相乙腈的体积分数分别为10%、30%和50%,查看10种目标物的分离度、洗脱速度和峰形等情况,结果表明选择以初始乙腈体积分数为30%最为合适。
分别选择纯水、乙腈-纯水(1∶1)作为样品定容液,对同一样品2倍稀释后进行对比测试。结果表明,用纯水和乙腈-纯水(1∶1)时10种目标物的保留时间和灵敏度的重现性都很好,纯水做定容液时响应值略低,但能完全满足检测要求,因此选择纯水作为样品定容液。生活饮用水较为干净,样品经0.22 μm滤膜可以直接进样测定,无需有机溶液稀释,这样预处理方式最为简便高效。
综合考虑本次试验:以甲酸-水(0.1∶99.9)溶液和乙腈作为流动相,采用梯度洗脱程序;水相比例由高到低,使目标物充分保留。能够使10种农药最大程度地分离,同时,含有低浓度甲酸的溶剂对电喷雾离子化较为有利,通过多反应监测色谱图能较好定性、定量分析10种农药。10种目标物在0~4 min内较快出峰且有效分离,峰形尖锐、对称性好,各峰附近无杂峰。梯度洗脱程序见表1。
2.2 质谱条件优化
采用仪器自带软件IntelliStart™和Quanpedia™功能,将农药混合标准溶液进行调谐和进样分析,对锥孔电压、碰撞气能量、碰撞气流量等进行优化,使分子离子与特征碎片离子对信号强度的响应值达到最佳,质谱条件见表2。50 μg/L的10种农药混合标准溶液的提取离子流色谱图见图1。
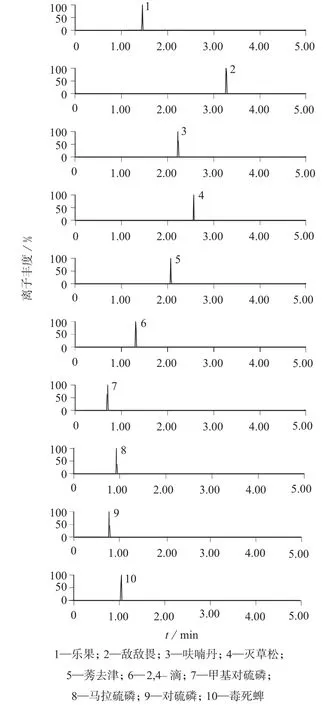
图1 10种农药混合标准溶液提取离子流色谱图
2.3 线性方程和检出限
将混合标准溶液稀释至质量浓度分别为0.5,2.5,5,10,25,50 μg/L,以各目标化合物的质量浓度为横坐标,以定量离子对应的色谱峰面积为纵坐标绘制标准工作曲线,10种化合物的质量浓度在0.5~50 μg/L范围内呈线性关系,计算各化合物的线性回归方程和相关系数,以3倍信噪比(3S/N)计算方法的检出限。线性方程、相关系数、检出限数据见表3。由表3可知,10种农药检出限均满足GB 5749-2006 《生活饮用水卫生标准》的限值要求。
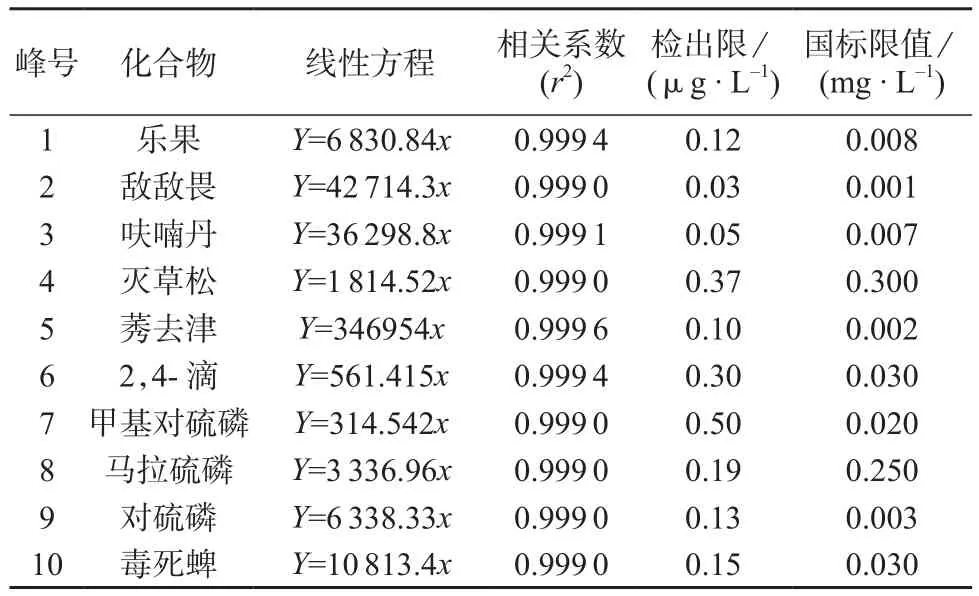
表3 线性方程、相关系数和检出限
2.4 加标回收试验
向纯水基质中分别加入2.5,10,50 μg/L 3个浓度水平的10种农药混合标准溶液,分别测定6次计算相对标准偏差和回收率,结果见表4。
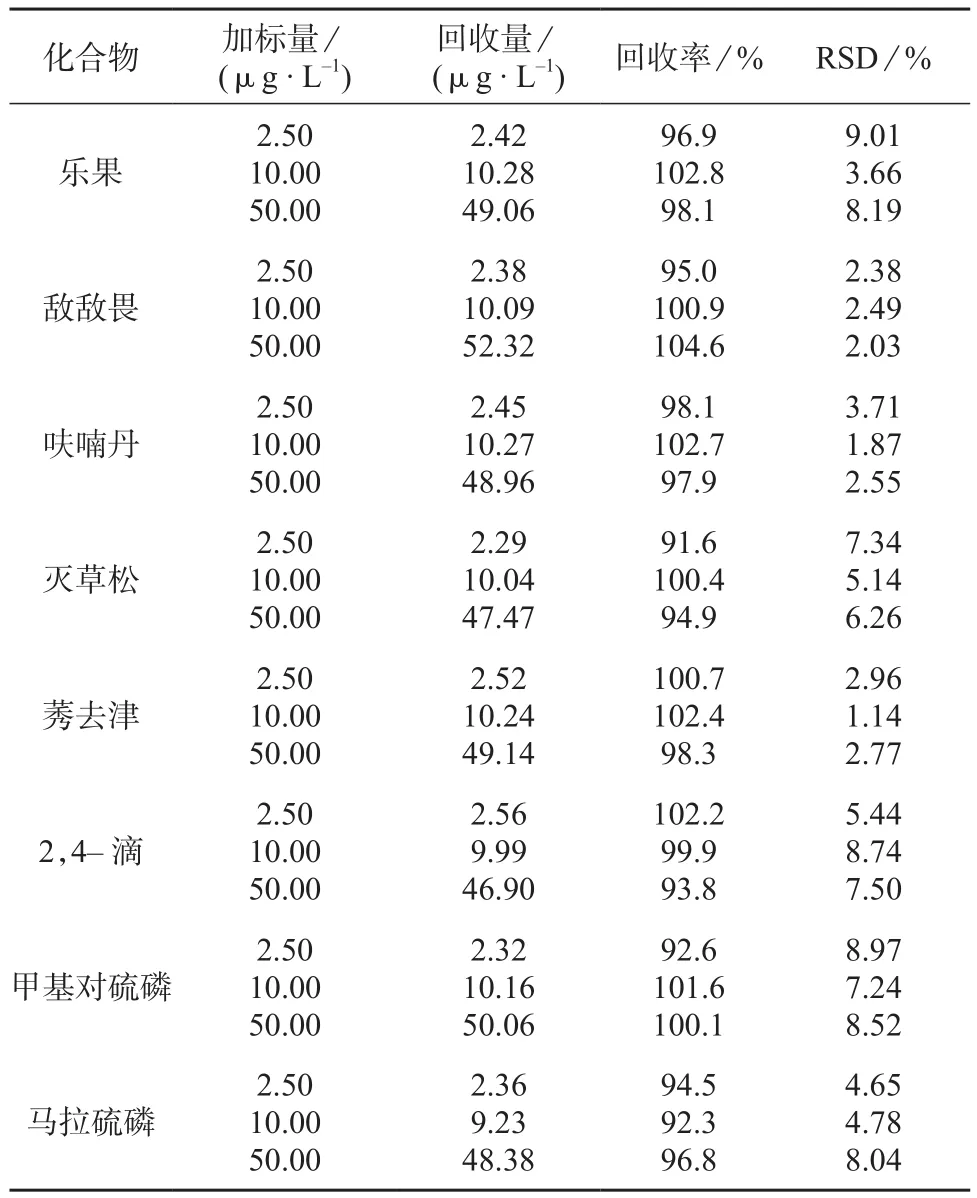
表4 精密度和回收试验结果(n=6)
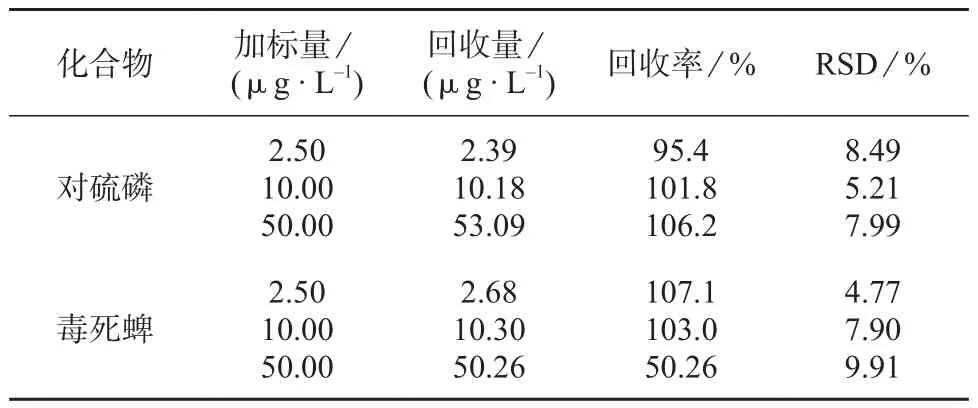
续表4
3 结语
采用超高效液相色谱-串联质谱法同时测定生活饮用水中10种农药残留量,可快速有效地分离待测目标物。该法前处理简单,灵敏度高、精密度和准确度好,满足生活饮用水全分析的实际检测需要。
[1] GB 5749-2006 生活饮用水卫生标准[S].
[2] 胡晓科,冯波,杨晓松.固相萃取-高效液相色谱法同时检测生活饮用水中的呋喃丹、莠去津、微囊藻毒素-LR[J].中国卫生检验杂志,2015(11): 1 699-1 702.
[3] GB/T 5750.9-2006 生活饮用水标准检验方法农药指标[S].
[4] 王雷,张艳霞.水体中有机磷残留的分析检测进展[J].环境研究与监测,2014(3): 70-74.
[5] 王正芳,宋维涛,宋晓娟,等.分散液液微萃取-高效液相色谱法测定环境水样中三嗪类农药[J].污染防治技术,2014,27(3):72-75.
[6] 李凌,张付刚,李建,等.固相萃取-气质联用测定水中23种有机氯有机磷农药污染物[J].环境卫生学杂志,2014,4(3):305-309.
[7] 高玲,罗晓飞,卢丹,等.圆盘膜萃取-气相色谱-串联质谱法快速测定水中40种有机磷农药[J].卫生研究,2013,42(6):1 062-1 066.
[8] 宋伟,林姗姗,孙广大,等.固相萃取-气相色谱-质谱联用同时测定河水和海水中87种农药[J].色谱,2012,3(3): 318-326.
[9] 郑磊,胡小键,张海婧,等.饮用水中痕量灭草松和2,4-滴的固相萃取-高效液相色谱串联质谱测定法[J].环境与健康杂志,2015,32(3): 246-2485.
[10] 赵慧琴,刘斌.水中莠去津等11种农药残留的超高效液相色谱/串联质谱分析法[J].职业与健康,2015,31(9):1 179-1 181.
[11] 郭忠,段江平,王妍妍,等.液相色谱串联质谱法直接进样测定生活饮用水中呋喃丹、莠去津、灭草松和2,4-滴[J].中国卫生检验杂志,2013,23(5): 1 132-1 134.
[12] 康莉,刘红河,刘桂华,等.LC/MS/MS测定饮用水和水源水中五氯酚、灭草松、草甘膦和2,4-滴[J].中国卫生检验杂志,2013,23(14): 2 871-2 873.
[13] 单晓梅,谢继安,沈登辉,等.水中灭草松、莠去津、2,4-滴、呋喃丹和微囊藻毒素UPLC-MS-MS快速测定方法[J] .环境卫生学杂志,2015,4(2): 178-182.
[14] 付慧,张海婧,胡小建,等.水中呋喃丹及5种有机磷农药的超高效液相色谱串联质谱测定法[J].环境与健康杂志,2015,32(3): 243-245.
[15] 吴春英,谷风,白鹭,等.固相萃取-超高效液相色谱-三重四级 杆质谱联用仪同时测定水中有机磷农药残留[J].中国卫生检验杂志,2013,23(5): 1 132-1 134.
[16] 朱月芳,尹燕敏,秦宏兵.直接进样-超高效液相色谱串联质谱法同时测定水源水中9种农药[J].化学分析计量,2016,25(1): 19-21.
[17] 孟凡飞,田葆萍,张光友,等.液质联用技术在水质检测中的应用研究进展[J].职业与健康,2014(16):2 344-2 346.
Determination of 10 Pesticide Residues in Drinking Water by UHPLC-MS
Zhu Jianfeng, Chen Jun, Miu Ying
(Jiangyin Center for Disease Control and Prevention, Jiangyin 214400, China)
A method of UHPLC-MS/MS was used for the determination of 10 pesticide residues in drinking water. The water samples were directly injected,then separated on a Waters ACQUITY UPLC®BEH C18column with the mixture of acetonitrile-0.1% formic acid solution as mobile phase for gradient elution. MRM was adopted for analysis in MS detection. The linear relationship was found between the peak area and the mass concentration of 10 pesticide was in the range of 0.5-50 μg/L with detection limit (3S/N)of 0.03-0.50 μg/L, the recovery rates were in the range of 91.6%-107.1% with RSD in the range of 1.14%-9.91%. This method is simple and accurate, and it can be applied to the determination of pesticide residues in drinking water.
UHPLC-MS/MS; drinking water; pesticide residues
0657.63
1008-6145(2016)05-0095-04
10.3969/j.issn.1008-6145.2016.05.025
联系人:朱建丰;E-mail: 1036005831@qq.com
2016-07-12
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
食品安全导刊(2021年21期)2021-08-30
理化检验-化学分册(2020年12期)2021-01-26
新世纪智能(英语备考)(2018年11期)2018-12-29
食品界(2018年8期)2018-09-03
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
环境科技(2016年2期)2016-11-08
中国环境监察(2016年7期)2016-10-23
中国现当代社会文化访谈录(2016年0期)2016-09-26
