
DNA聚合酶基因在范可泥贫血症家系突变状态中的影响研究
2016-10-13 09:08刘宇王培昌
河北医药 2016年19期
刘宇 王培昌
·论著·
DNA聚合酶基因在范可泥贫血症家系突变状态中的影响研究
刘宇王培昌
目的通过对范可泥贫血症(fanconi anemia,FA)家系DNA聚合酶基因突变分析,初步观察其稳定突变位点,为上述疾病易感基因风险突变位点的进一步确定奠定基础。 方法FA家系15例成员作为研究对象对家系先征者DNA聚合酶基因家族POLA、POLB、POLD1、POLD2、POLE、POLG测序,检测突变位点,并分析筛选有义突变位点;对家系成员在先征者有义突变区域直接测序。结果FA患儿DNA聚合酶的外显子共有四个突变位点。其中,POLD1 19p13.3-1 3.4 EXON2 50403975 G>A,密码子由CGU变为CAU,氨基酸由精氨酸变为组氨酸;POLE1 12p24.3 EXON43-44 132688389 A>G,密码子由GAG变为GGG,编码氨基酸由谷氨酸变为甘氨酸;家系中患儿的母亲、外祖父发生POLD1 EXON2 50403975 G>A;家系中患儿的外祖母、外祖父、祖母、父亲、母亲、二伯、四伯、大姑、二姑、两个堂姐均发生了POLE1 EXON43-44 132688389 A>G。结论通过对FA家系成员在上述已经筛选出的两个突变位点进行测序,该家族成员广泛携带这两个突变位点。这两个基因的突变位点应得到足够的重视,其可能为FA疾病的稳定突变位点。
范可尼贫血症;共济失调毛细血管扩张症;DNA聚合酶基因;家系;基因突变;基因表达
范可泥贫血症(FA)是一种常染色体或X染色体连锁的隐性遗传病,其又称为范可尼综合征,患者具有先天性发育异常、骨髓衰竭、高度癌症易感性等特征。FA在人群中发病比例十分罕见,仅为1∶1 000 000~5∶1 000 000[1]。1927年由瑞士Fanconi[2]首先命名并报道。FA患者的临床表现具有多样性的特点,多伴有FA患者可出现发育迟滞现象, 表现为体格矮小、小头畸形、眼球小,皮肤色素沉着,拇指或桡骨不发育或缺如,呈多指、有指蹼等,还可合并贫血、出血、感染。FA患者多数伴有进行性骨髓衰竭,最终约90%的FA患者死于此原因[3]。患者还好发肿瘤,25%的FA患者以实体肿瘤而就诊[3],其中多见鳞状细胞癌(食管癌、阴道癌)和头颈部癌,此外还有脑瘤、星状细胞瘤、肝癌、肾母细胞癌和乳腺癌等[4]。近年来对FA的研究进展快速,现临床将FA分为15 个亚型,并已有15 个相关基因相应被克隆,且发现这15个基因中任何一个特定突变或缺失均与不同的FA亚型相对应,呈现不同的临床特征,这些基因均参与DNA交联损伤修复[5-8]。进一步研究发现,在大多数情况下,FA是由FA基因中的一个发生突变而不是多个发生突变导致,已经确定, 这15个FA 基因的突变导致超过95%已知患者的FA 综合征。然而,在实际病例中还发现有一些FA患者并没有已知的15个FA 基因的突变,说明还有一些未知的FA 相关基因有待进一步发现。众所周知,真核细胞主要有5种DNA聚合酶,分别为DNA聚合酶α(DNA polymerase α,polα)、DNA聚合酶β(DNA polymerase β, polβ)、DNA聚合酶γ(DNA polymerase γ, polγ)、DNA聚合酶δ(DNA polymerase δ, polδ)、DNA聚合酶ε(DNA polymerase ε, polε)。DNA聚合酶在DNA复制、损伤修复、线粒体DNA复制等均起着十分重要的作用。本文检测了一个FA家系编码DNA polα、β、γ、δ、ε的部分基因序列,以期发现FA家系稳定的突变位点,为滞后FA风险突变位点的筛选及机制研究提供一定的基础。
1 资料与方法
1.1一般资料FA家系,家系图见图1。
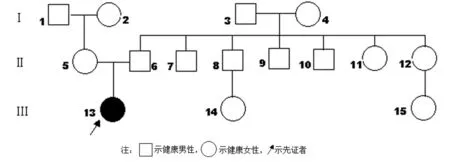
图1本研究FA家系图谱,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15分别代表外祖父、外祖母、祖父、祖母、母亲、父亲、大伯、二伯、三伯、四伯、大姑、二姑
1.2仪器与试剂仪器、试剂和耗材各称生产厂家见表1、2。

表1 主要仪器

表2 主要试剂、耗材
1.3方法
1.3.1标本采集和处理:征得家系成员知情同意的情况下,当地采集家系成员14人的外周血5 ml置于乙二胺四乙酸(Ethylenediaminetetraaceticacid,EDTA)抗凝管中,干冰保存运输回实验室。
1.3.2全血DNA提取:详见试剂盒说明书。
1.3.3引物设计:对所研究的六条基因POLA、POLB、POLD1、POLD2、POLE、POLG外显子设计引物,引物设计采用Primer Premier 5.0专业软件,生工生物合成。
1.3.4PCR扩增:采用普通聚合酶链式反应(PCR)PCR技术,直接采用设计引物对上述六条基因的外显子进行扩增。见表3。

表3 PCR扩增体系及参数
注:退火温度根据引物TM值设定,延伸时间根据片段长度设定
1.3.5PCR产物纯化:PCR产物电泳后,用DNA纯化试剂盒纯化,详见试剂盒说明书。
1.3.6PCR产物测序:将上述已纯化好的PCR产物交美吉生物医药科技有限公司测序。
1.3.7患儿目的基因序列分析:将上述测序结果用Chromas软件与美国NCBI基因库所公布的POLA、POLB、POLG、POLD1、POLD2和POLE六条基因序列进行比对,寻找突变位点。发现突变位点后对该位点反向测序进行验证。
1.3.8家系成员突变位点位置测序:提取家系成员基因组DNA,对家系成员在上述比对结果所查找出的有意义的突变位点位置进行PCR扩增、纯化、测序。
2 结果
2.1患儿目的基因突变位点信息对该FA家系先证者POLA、POLB、POLG 、POLD1、POLD2和POLE六条基因外显子序列进行比对。发现突变位点后对该位点所在六个基因反向测序发现,外显子区共有4个突变位点,内含子区共发现7个突变位点。其中有两个外显子区位点突变导致了氨基酸改变,分别是位于POLD1基因 19 p13.3~13.4 EXON2 50403975,密码子由CGU突变为CAU,编码氨基酸由精氨酸变为组氨酸;另一突变位点位于POLE1基因12 p24.3 EXON43-44 13268838,密码子由GAG突变为GGG,编码氨基酸由谷氨酸变为甘氨酸。见表4、5,图2、3。
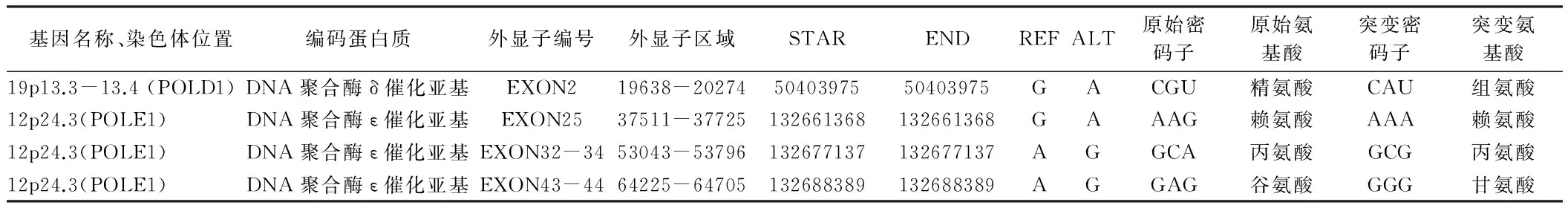
表4 FA患儿DNA聚合酶基因外显子突变位点分析
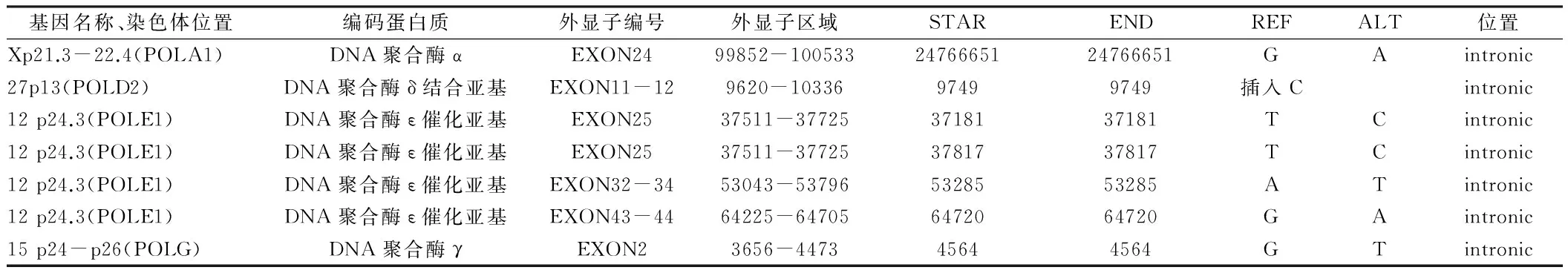
表5 FA患儿DNA聚合酶基因非外显子区域突变位点分析
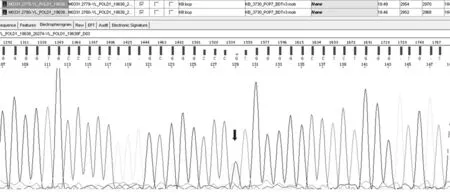
图2 DNA POLD1EXON2部分测序结果;箭头示POLD1 EXON250403975突变(G>A)
2.2家系成员在患儿突变位点位置测序结果分析POLD1 EXON2 50403975与POLE1 EXON45 132688389均导致了氨基酸的改变,因突变位点分别位于DNApol δ催化亚基和DNA pol ε催化亚基,推测此两个突变位点对DNA pol δ、DNA pol ε功能影响较大,为分析其是否为该家系稳定遗传突变位点,本研究对家系其他成员该位点进行测序。发现家系中患儿的母亲、外祖父亦见POLD1 EXON2 50403975G>A;家系中患儿的外祖母、外祖父、祖母、父亲、母亲、二伯、四伯、大姑、二姑、两个堂姐均见POLE1 EXON43-44 132688389 A>G。见表5、6,图4、5。
3 讨论
FA是由于基因损伤修复缺陷所致,而DNA损伤修复是一复杂的生理过程,与DNA碱基错配、氧化损伤、交联、插入或缺失等的识别、切除及修复密切相关,那么,DNA聚合酶家族与FA关系如何,是否DNA聚合酶基因突变直接或间接导致了FA的发生,笔者目前尚未见报道。FA为一种罕见的常染色体或X染色体连锁隐性遗传病,一般5~10年发病[9]。有研究报道,除去骨髓增生异常综合征(myelodysplastic syndromes,MDS)和急性髓细胞白血病(acute myelocytic leukemia,AML)所占比例,FA患者患实体肿瘤的累积发病率已达到76%[10]。FA患者的临床表现有些仅表现为轻微贫血,部分表现为多脏器或身体特定部位发育畸形伴严重的再生障碍性贫血(aplastic anemia,AA),有些以AML为首发表现。经对FA患者临床表现总结,此病的临床表现个体差异很大,除临床表现及常规检查应作为此病诊断的重要标准,还要结合其他诊断方法,如基因诊断等。
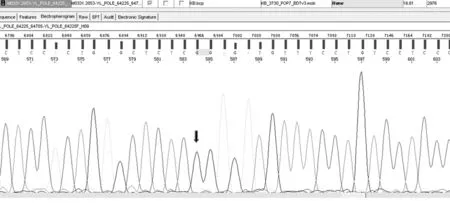
图3 DNA POLE1 EXON45部分测序结果;箭头示POLE1 EXON45132688389突变(A>G)
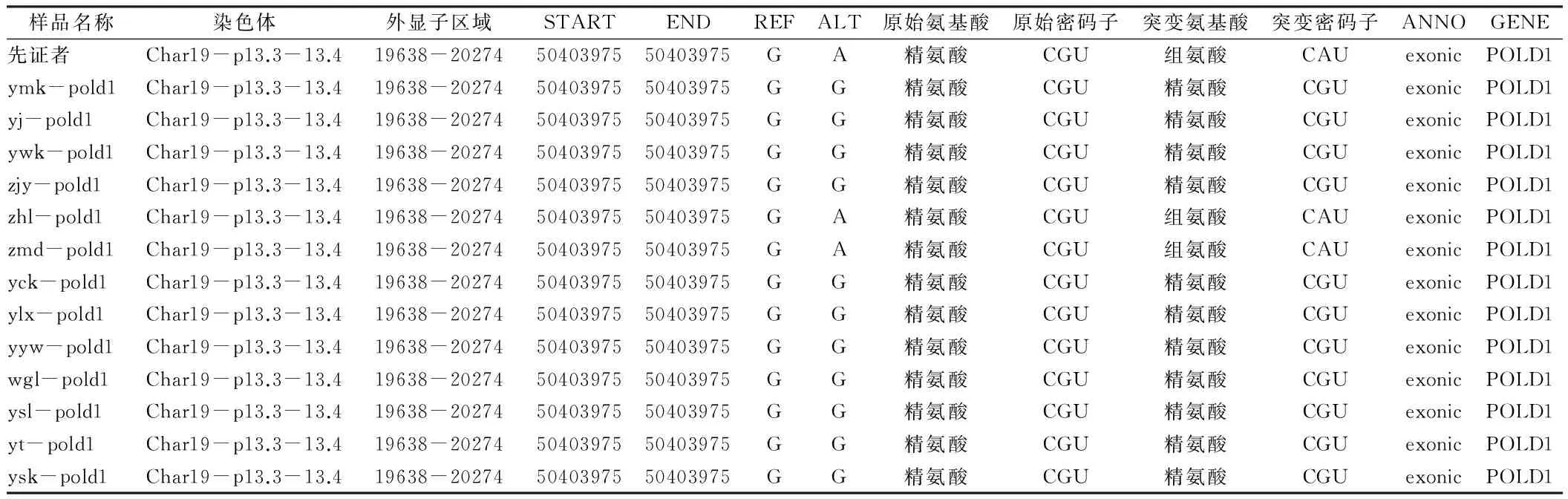
样品名称染色体外显子区域STARTENDREFALT原始氨基酸原始密码子突变氨基酸突变密码子ANNOGENE先证者Char19-p13.3-13.419638-202745040397550403975GA精氨酸CGU组氨酸CAUexonicPOLD1ymk-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1yj-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1ywk-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1zjy-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1zhl-pold1Char19-p13.3-13.419638-202745040397550403975GA精氨酸CGU组氨酸CAUexonicPOLD1zmd-pold1Char19-p13.3-13.419638-202745040397550403975GA精氨酸CGU组氨酸CAUexonicPOLD1yck-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1ylx-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1yyw-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1wgl-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1ysl-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1yt-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1ysk-pold1Char19-p13.3-13.419638-202745040397550403975GG精氨酸CGU精氨酸CGUexonicPOLD1
注:ymk、yj、ywk、zjy、zhl、zmd、yck、ylx、yyw、wgl、ysl、yt、ysk分别代表患儿的父亲、四伯、大伯、外祖母、外祖父、母亲、二伯、大姑、祖父、祖母、二姑、姐姐、三伯
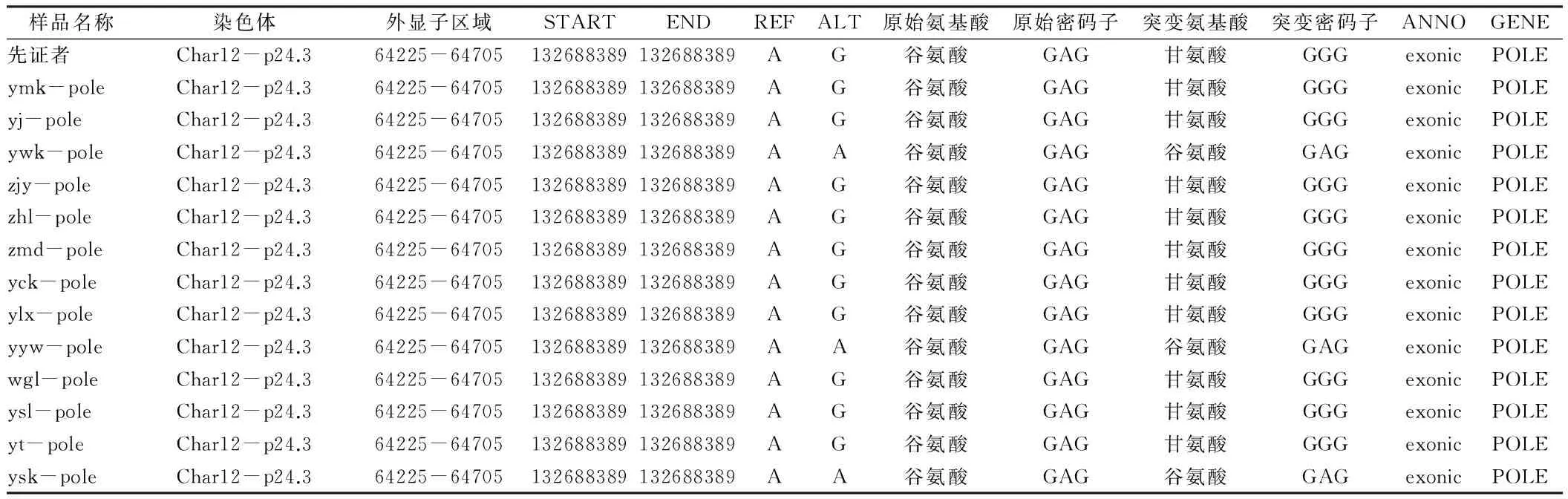
表6 POLE EXON45132632393A>G在家系成员中突变情况位点分析
注:ymk、yj、ywk、zjy、zhl、zmd、yck、ylx、yyw、wgl、ysl、yt、ysk分别代表患儿的父亲、四伯、大伯、外祖母、外祖父、母亲、二伯、大姑、祖父、祖母、二姑、姐姐、三伯
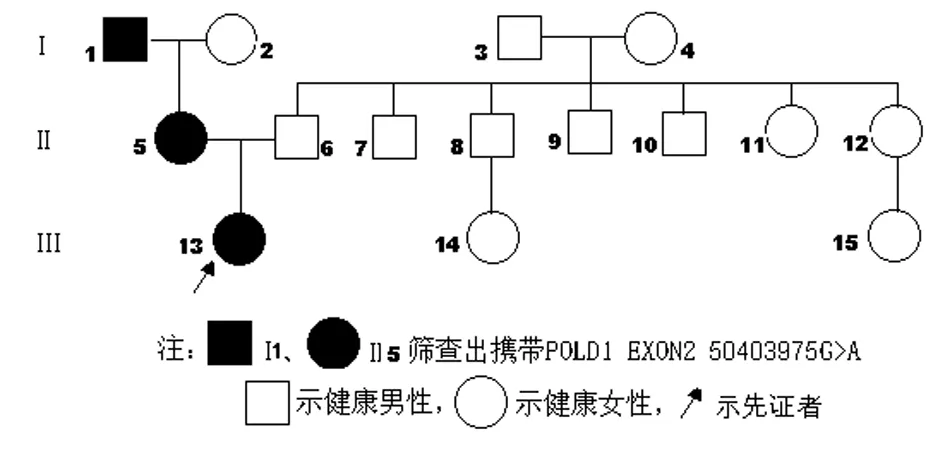
图4FA家系POLD1 EXON2 50403975突变位点携带示意图.1、2、3、4、5、6、7、8、9、10、11、12、14、15分别代表患儿的外祖父、外祖母、祖父、祖母、母亲、父亲、大伯、 二伯、三伯、四伯、大姑、二姑、患儿、大堂姐、二堂姐
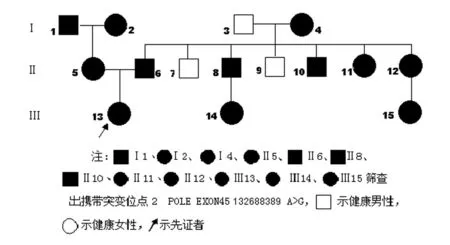
图5FA家系POLE1 EXON45 132688389突变位点携带示意图. 1、2、3、4、5、6、7、 8、9、10、11、12、14、15分别代表患儿的外祖父、外祖母、祖父、祖母、母亲、父亲、大伯、二伯、三伯、四伯、大姑、二姑、患儿、大堂姐、二堂姐
大量研究显示,FA发病与一系列参与DNA 交联损伤修复基因的突变有关,迄今已克隆了15个FA基因,它们是FANC A,FANC B,FANC C,FANC D1,FANC D2,FANC E,FANC F,FANC G,FANC I,FANC J,FANC L,FANC M,FANC N,FANC O,FANC P[11-14]。且已明确95%的FA患者是由上述FA基因中的某一个发生突变所致[15]。但是仍有少部分FA病例突变未发生在上述区域,提示尚有一些未知的基因突变与FA发生有关联[1]。
真核DNA聚合酶家族在基因的损伤修复中起到至关重要的作用。DNApol α的生物学功能包括复制(引发复制起始)、DNA双链断裂修复(DNA double strand breaks repair,DSBR)、端粒稳定性维持、S 期关卡调控;DNA pol β主要参与DNA损伤修复;DNA pol γ主要参与线粒体DNA 复制;DNA pol δ除参与DNA复制等,还参与DNA损伤修复,包括错配修复(Mis-match repair,MMR)、碱基切除修复(Base excision repair,BER)、核苷酸切除修复(Nuleotide excision repair,NER)、DSBR、重组、端粒稳定性维持、S 期关卡调控等作用;DNA polε的生物学功能主要为复制、损伤修复(MMR、BER、DSBR)、重组、转录沉寂、S 期关卡调控[16]。此外,DNA聚合酶与肿瘤易感性有着密不可分的联系,已有研究发现原发性肝癌组织中DNA pol δ催化亚基p125表达水平较癌旁组织高,肺癌组织中DNA pol δ第4个亚基p12 低表达,也有发现结肠癌组织及细胞系中发现DNA pol δ催化亚基p125 的编码基因POLD1 发生突变[17]。
DNA pol δ是最保守的真核生物DNA 聚合酶,POLD1编码 DNA聚合酶δp125催化亚基,其位于19号染色体q13.3~13.4上。POLD1的cDNA全长3 443 bp,包含了27个外显子和26 个内含子,该片段可编码1 107个氨基酸[17]。DNA聚合酶δ参与真核生物DNA合成、损伤、修复和细胞周期调控等过程,与肿瘤的发生有着密不可分的关系,它是非常重要聚合酶之一。真核DNApol ε有4个亚基,分别是Pol2(最大亚基、催化亚基、A亚基)、Dpb2、Dpb3 和Dpb4。其中催化亚基在DNA复制、校正、损伤修复等起着十分重要的作用。Pol2 的N端结构域不但有聚合酶活性和还具有3’-5’外切酶活性。起着催化活性作用的C端结构域不仅介导催化亚基与另外3个小的调节亚基间的相互作用,而且与S 期的复制关卡装置相关联[18]。有研究显示, 鼠双微基因2(murine double minute 2,MDM2)是肿瘤抑制因子p53 的一种调节因子。MDM2 可与DNA聚合酶ε催化亚基的C 端结构域相结合[19],通过诱导使DNA聚合酶ε结构发生改变, 对DNA损伤作出反应[20]。DNA聚合酶δ、DNA聚合酶ε是真核生物两种非常重要的酶,研究表明DNA聚合酶δ突变基因型的出现会提高肿瘤易感性[21],DNA聚合酶ε的催化亚基Pol2 与其他真核DNA 聚合酶B家族成员尤其是DNA聚合酶δ催化亚基相比结构有同源性(图6[22])。
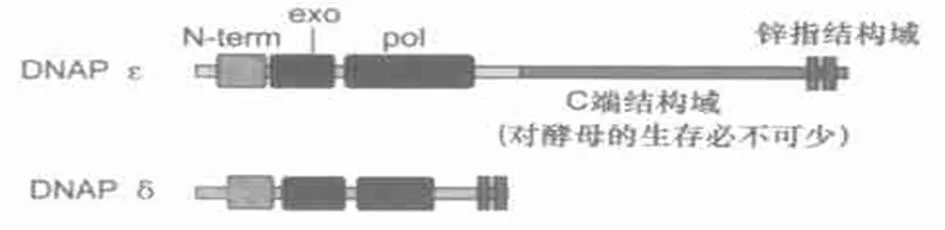
图6 DNAPE与DNAPD催化亚基的结构比较
本研究病例来自内蒙古自治区巴彦淖尔地区1例EA家系,该家系共3代14名成员。先证者,女,11岁,口唇发白,明显贫血貌,手部拇指有特征性的先天畸形,身材比同龄孩子矮小,有儿童MDS血象表现。先症者于2009年在首都医科大学儿童医院血液科就诊,接受临床体征和骨髓穿刺检查,诊断为范可尼贫血,询问其他家系成员均无此病。
对患儿DNA聚合酶基因测序,在外显子区域有4处突变位点,内含子区共7个突变位点。其中两处有义外显子突变位点,分别是POLD1 EXON2 50403975(G>A),家系中患儿的母亲、外祖父在该位点亦有突变;POLE1 EXON43-44 132688389(A>G),家系中患儿的外祖母、外祖父、祖母、父亲、母亲、二伯、四伯、大姑、二姑、两个堂姐亦有该处突变。患儿非外显子区域的(内含子)突变位置多位于POLE1 12 p24.3。
POLD1 EXON2 50403975(G>A)突变,密码子由CGU变为CAU,编码的氨基酸由精氨酸变为组氨酸,该突变位于DNA pol δ催化亚基;POLE1 EXON43-44 132688389(A>G),密码子由GAG变为GGG,编码氨基酸由谷氨酸变为甘氨酸,该突变位于DNA pol ε催化亚基。上述2个突变可能导致了DNA聚合酶DNA复制及损伤修复功能的改变,推测与先证者发病存在一定关联。尤其引人关注的是,该家系中发生上述两处突变位点的成员应密切回访,观察是否发病。
本研究还存在很多不足,由于疾病本身的罕见性导致了研究例数过少,虽在研究中发现了该FA家系中存在的潜在的易感突变位点,但要最终确定还需在更多家系中验证。此外,还需要继续对DNA聚合酶基因家族扩大测序范围,才能更全面地找到DNA聚合酶基因对该病的易感突变位点。DNA聚合酶对FA的发生发展以及对其发病机制有何作用还有待更加深入的研究。
该部分对一个FA家系成员进行了DNA聚合酶基因测序,在先证者中发现两处有义外显子突变位点POLD1 EXON2 50403975(G>A)、POLE1 EXON43-44 132688389(A>G)。在未发病的家系成员中患儿的母亲、外祖父亦有POLD1 EXON2 50403975(G>A);患儿的外祖母、外祖父、祖母、父亲、母亲、二伯、四伯、大姑、二姑、两个堂姐亦有POLE1 EXON43-44 132688389(A>G)。
1阮春燕,韩金花,刘婷,等.范可尼贫血症与DNA交联损伤修复.中国科学杂志,2014,44:387-396.
2Fanconi G.Familire infantile perniziosaartige Anmie(pernizioses Blutbild and Konstitution).Jahrb Kinderh,1927,117:257-280.
3张莹莹,吴占河.范可尼贫血的新概念和临床与实验室诊断.诊断学理论与实践,2013,12:501-503.
4Alter B. Fanconi Anemia.NCBI Bookshelf. A service of the National Library of Medicine, National Institute of Health,2011.1-31.
5Kim H,D’Andrea AD.Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev,2012,26:1393-1408.
6Moldovan GL,D’Andrea AD.To the rescue: the Fanconi anemia genome stability pathway salvages replication forks.Cancer Cell,2012,22:5-6.
7D’Andrea AD.The Fanconi anemia/BRCA signaling pathway:disruption in cisplatin-sensitive ovarian cancers.Cell Cycle,2003,2:290-292.
8Wang W.Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins.Nat Rev Genet,2007,8:735-748.
9岳燕,罗荣牡,司英健.外周血造血干细胞移植联合间充质干细胞输注治疗范可尼贫血患儿1例并文献复习.中国小儿血液与肿瘤杂志,2015,20:139-144.
10Alter BP.Cancer in Fanconi anemia,1927-2001.Cancer,2003,97:425-440.
11Kim H,D’Andrea AD.Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway.Genes Dev,2012,26:1393-1408.
12Moldovan GL,D’Andrea AD.To the rescue: the Fanconi anemia genome stability pathway salvages replication forks.Cancer Cell,2012,22: 5-6.
13D’Andrea AD.The Fanconi anemia/BRCA signaling pathway: disruption in cisplatin-sensitive ovarian cancers.Cell Cycle,2003,2:290-292.
14Wang W.Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet,2007,8:735-748.
15Park JW, Pitot HC, Strati K, et al. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer.Cancer Res,2010,70: 9959-9968.
16鲍羿,杨克恭,洪斌.真核DNA聚合酶ε.生命的化学杂志,2004,24:523-526.
17阮细玲,吴琼,黄怡,等.DNA 聚合酶δ与肿瘤的关系.临床与实验病理学杂志,2012,28:1375-1377.
18Pospiech H,Syväoja JE,et al.DNA poltmerase epsilon-nore than a polymerase.Scientific World Journal,2003,3:87-104.
19Vlatkovic N,Fuerrera S,Li Y,et al.MDM2 interacts with the c-terminus of the catalytic subunit of DNA prelymerase epsilon.Nucleic Acids Res,2000,28:3581-3586.
20Joenje H,Obstra AB,Wijker PC,et al.Evidence for at least 8 Fanconi anemia genes.Am J Hun Cenet,1997,61:940-944.
21Buermeyer AB,Deschenes SM,Baker SM,et al.Mammalian DNA mismatch repair.Annu Rev Genet,1999,33: 533-64.
22Hubscher U,Nasheuer HP,Syväoja JE,et al.Eukaryotic DNA polymerases,growing famiy.Trends Bi ochem Sci ,2000,25:143-147.
Study on the mutation status of DNA polymerase genes in Fanconi anemia family constellation
LIUYu,WANGPeichang.
DepartmentofClinicalLaboratory,XuanwuHospitalAffiliatedtoCapitalMedicalUniversity,Beijing100153,China
ObjectiveThrough analysing the mutation status of DNA polymerase genes in Fanconi anemia (FA) family constellation to observe initially its stable mutation sites in order to establish basis further for determining the risky mutation site of predisposing genes of the disease.MethodsThe 15 family menbers of FA family constellation were enrolled in the study. DNA polymerase gene family members including POLA,POLB,POLD1,POLD2,POLE and POLG of FA proband were analyzed by gene sequencing to screen meaningful mutation sites,and DNA of the family members was analyzed by direct sequencing in plus sense mutation region of the proband.ResultsThere were four mutation sites in exon of DNA polymerase in children patients with FA in which POLD1 19p13.3~13.4 EXON2 50403975 G>A,the codon was changed from CGU to CAU,and coding amino acid was changed from glutamic acid to glycine,moreover, POLE1 12p24.3 EXON43-44 132688389 A>G, the codon was changed from GAG to GGG,and coding amino acid was changed from glutamic acid to glycine,besides, POLE1 12p24.3 EXON43-44 132688389 G>A in patient's mother and adoptive grand-father,and there appeared POLE1 EXON43-44 132688389 A>G in patient's adoptive grand-mother,adoptive grand-father, grandmother,father,mother,the second uncle,the 4th uncle,the first aunt,the second aunt,two sisters in-law.ConclusionThrough the sequencing for two gene mutation sites of FA family members that have been selected, it is indicated that the family members take along with the two gene mutation sites generally.Thus,the two gene mutation sites should be payed much attention to,besides,which may be the stable mutation sites of FA.
Fanconi anemia;ataxiatelangiectasia;DNA polymerase genes; family constellation; gene mutation; gene expression
10.3969/j.issn.1002-7386.2016.19.001
100153北京市,首都医科大学宣武医院检验科
王培昌,100153北京市,首都医科大学宣武医院检验科;
E-mail:583494898@qq.com
R 394.112
A
1002-7386(2016)19-2885-06
2016-03-02)
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
新作文·小学高年级版(2022年3期)2022-04-01
意林·少年版(2020年22期)2020-12-10
启迪与智慧·下旬刊(2019年5期)2019-09-10
吉林林业科技(2018年6期)2018-11-21
现代检验医学杂志(2016年1期)2016-11-12
中外医疗(2015年11期)2016-01-04
听力学及言语疾病杂志(2015年5期)2015-12-24
中国当代医药(2015年30期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
