
无锡城乡年轻居民肠道菌群多样性研究
2016-10-11 07:31:34乔健敏张家超侯强川黄卫强霍冬雪张和平
食品与生物技术学报 2016年8期
乔健敏, 张家超, 郑 艺, 侯强川, 黄卫强, 霍冬雪, 郭 壮, 张和平
(内蒙古农业大学 乳品生物技术与工程教育部重点实验室,内蒙古 呼和浩特 010018)
无锡城乡年轻居民肠道菌群多样性研究
乔健敏, 张家超, 郑 艺, 侯强川, 黄卫强, 霍冬雪, 郭 壮, 张和平*
(内蒙古农业大学 乳品生物技术与工程教育部重点实验室,内蒙古 呼和浩特 010018)
采用PCR和DGGE相结合的技术,研究了生活于无锡城市和乡村的青年志愿者肠道菌群的多样性。基于DGGE指纹图谱,使用聚类和PCA分析对志愿者肠道微生物相似性进行分析,使用Shannon-Weine多样性指数(H)、丰度(S)和均匀度(EH)对志愿者肠道微生物多样性进行分析,对图谱中具有代表性的共性和特异性条带进行胶回收和测序以分析志愿者肠道微生物组成;基于PCR技术在种水平上对城乡志愿者肠道内乳杆菌属 (Lactobacillus)和双歧杆菌属(Bifidobacterium)多样性进行定性分析。结果显示,无锡城乡青年居民间肠道微生物群落结构存在分开趋势,相似性小于城市或乡村居民内部;多样性方面差异不显著;不动杆菌属、双歧杆菌属、葡萄球菌属、乳杆菌属、梭菌属和瘤胃球菌属为城乡青年居民肠道内的共有菌属;乳杆菌属Lactobacillus(L.)和双歧杆菌属Bifidobacterium(B.)各类菌群在城乡居民肠道内分布差异不显著,其中L.plantarum,L.casei,L.salivarius和L.acidophilus以及B.longum,B.breve,B.animalis 和B.adolescentis分别为无锡青年居民肠道内的优势乳杆菌和双歧杆菌。综上分析,初步揭示了无锡城市和乡村青年居民肠道微生物多样性差异不显著。
无锡城乡年轻居民;微生物多样性;PCR技术;DGGE技术
17世纪50年代,在微生物学之父列文虎克借助自己发明的显微镜开始研究微生物的同时,关于人类肠道微生物的研究也随之开始[1-2]。据报道,健康成年人肠道微生物的总数达1014个,10倍于人体细胞,其微生物基因数量约为300万个,约为人类基因组基因数量的100倍。因此,研究者们将人体肠道微生物基因组称为人体的第二基因组[3]。这些微生物与宿主共进化,直接参与人体的能量代谢和免疫系统的调节,对于人的健康扮演着重要的角色[4]。
随着科学家使用不同的技术手段对不同年龄、不同国籍、不同遗传背景和生活习惯的人群肠道微生物的研究,发现人体的肠道微生物多样性在宿主2岁后开始趋于稳定并成人化,此后,随着宿主进入成人期,生活方式和饮食习惯成为影响宿主肠道微生物群落结构的更重要的因素[4-5]。早于1974年,Finegold[6]等通过对坚持传统日式饮食习惯的日本居民和已适应西方饮食习惯的日裔美国居民的肠道微生物进行研究,结果显示两类人群的肠道菌群结构明显不同。近些年,Wu等[7]通过对98个健康志愿者的肠道菌群进行研究,发现居民的肠道菌群可以划分为2个分别以拟杆菌属(Bacteroides)和普氏菌属(Prevotella)为核心的独立无交集的enterotype类型。enterotype类型的形成与长期的膳食有关,与性别以及肥胖等因素无关。Wu等进一步指出,长期食用高蛋白质高脂肪的食物能够促使肠道内拟杆菌属(Bacteroides)细菌的生长,而食用富含碳水化合物和单糖的食物则可以增加人肠道内普氏菌属(Prevotella)细菌的含量。
中国是一个陆地面积约960万平方千米的大国,不同地区具有不同的气候、文化和饮食习惯。而不同的人群具有不同的肠道菌群结构[8]。无锡地处中国东部长江三角洲地区,是传统的鱼米水乡,受吴文化的浸育,形成了典型的南方生活方式和饮食习惯,而城市和乡村因地理位置的不同生活方式也存在差异。
聚合酶链式反应-变性梯度凝胶电泳技术(PCR-DGGE)是一种不依赖于培养过程而对复杂微生物群落结构进行有效分析的分子生物学技术,因其具有低成本、可靠、可重复、操作方便等优点,近些年被广泛应用于微生物群落结构的研究[9]。Yoshida等[10]使用PCR-DGGE技术对太平洋、东海、东京湾以及荒川(Arakawa River)水环境中的放线菌进行评价,结果发现不同位置水环境中的放线菌群落结构不同,且同一水环境中不同深度微生物群落结构也不同。刘慧杰等[11]通过使用PCR-DGGE技术对红树林沉积物中细菌群落结构进行研究,证实PCRDGGE技术更能客观地反映红树林沉积物中真实的细菌群落结构信息,同时也证明红树林区域微生物多样性丰富,表明未知微生物资源的开发研究具有巨大的潜力。
本文作者应用PCR和DGGE相结合的技术对生活于无锡市区和乡村的青年居民肠道菌群多样性进行研究,并结合城乡居民生活环境和饮食习惯分析,探索了无锡城乡居民肠道菌群多样性的异同。
1 材料与方法
1.1 样本和试材
1.1.1 样本来源 选择16位居住于无锡城市和乡村、近一周内无出行记录的本地居民为研究对象,志愿者年龄均在19~35岁,身体质量指数(BMI)在18.5~25.0,近3个月内无抗生素类药物摄入,并签订知情同意书。同时,根据食品频率问卷调查(Food frequency questionnaire,FFQ)记录志愿者近3日膳食信息,并根据《中国食物成分表》计算志愿者脂肪、蛋白质和碳水化合物等营养成分的日摄入量[12],样品采集点分别为无锡乡村的滨湖区和城市的崇安区,各采样点间距离近100 km,具体样品信息见表1。
1.1.2 试验所用试剂 试验所用引物由上海桑尼生物科技有限公司合成,16S rRNA基因V3区扩增试剂均购于北京全式金生物技术有限公司,其它试验药品均为大连宝生物技术有限公司产品。
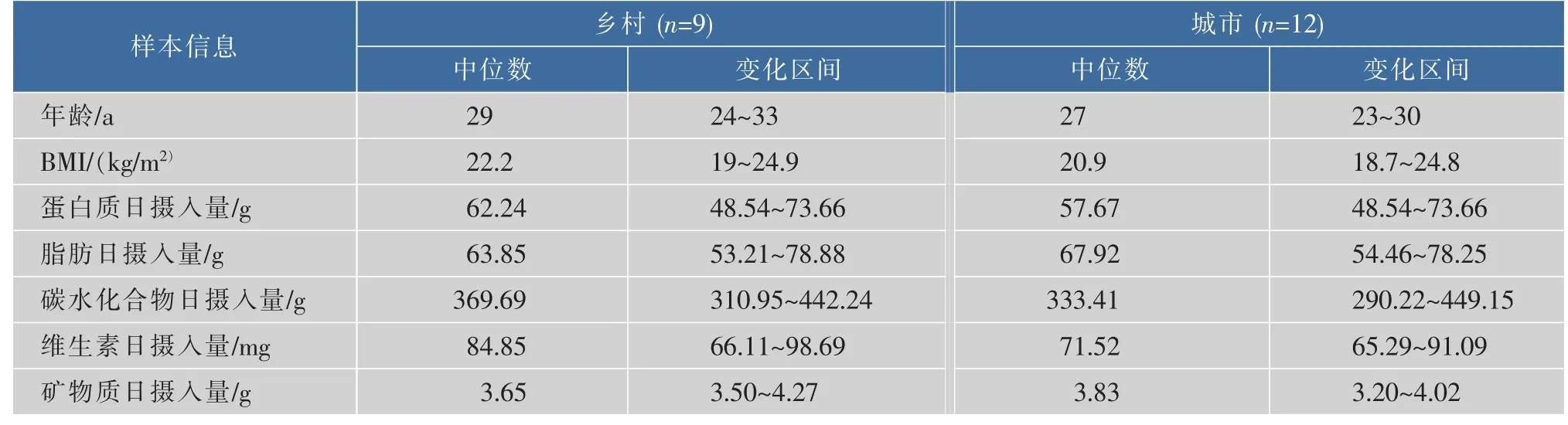
表1 样品信息Table1 Information of subjects
1.2 试验方法
1.2.1 粪便样品采集 早餐前分别采集志愿者新鲜粪便样品适量,放入采样管中,并立即加入适量保护剂(Takara Biotechnology,Dalian),粪便和保护剂体积比为1∶2,迅速在液氮中速冻后存放于冷冻环境,72 h内送回实验室于-80℃冷冻储存以备用。
1.2.2 粪便菌体基因组DNA提取 粪便菌体基因组 DNA的提取使用 QIAamp DNA stool mini kit (Qiagen)试剂盒[13]。得到的基因组DNA用ND-1000型微量紫外分光光度计测定浓度及A260/A280值,同时用1 g/dL的琼脂糖凝胶进行电泳检测,调整DNA质量浓度至ng/μL水平,于-20℃冷冻保存以用于后续试验。
1.2.3 基因组DNA 16S rRNA V3区PCR扩增 使用通用引物V3F+GC(5′-GCCGCCCGGGGCGCGCC CCGGGCGGCCCGGGGGCCTACGGGAGGCAGCAG-3′)和V3R(5′-ATTACCGCGGCTGCTGG-3′)进行16S rRNA基因V3区扩增,扩增片断长度为220 bp,所用仪器为Applied biosytems公司PCR仪。PCR反应体系 (50 μL)为:10×PCR缓冲液5 μL,dNTP (2.5 mmol/μL)4 μL,引物(10 pmol/μL)各1.5 μL,模板DNA(100 ng/μL)1.5 μL,Taq DNA聚合酶(5 U/μL)0.5 μL,ddH2O补足至50 μL。扩增反应程序为:95℃ 5 min;95℃ 1 min,55℃ 45 s,72℃1 min,30个循环;72℃7 min。得到的PCR产物用1 g/dL的琼脂糖凝胶电泳进行检测,其余置于-20℃冷冻保存以用于后续DGGE分析[14]。
1.2.4 粪便菌体基因组DNA变性梯度凝胶电泳(DGGE)分析 使用 DCode Universal Mutation Detection System(Bio-Rad)仪器,对16S rRNA基因V3区扩增产物进行DGGE分析。分析所用变性剂质量分数为8%,变性梯度为质量分数27%~52%,灌胶后电泳在60℃,200 V运行5 h。使用银染方法对所得胶图进行染色,拍照获得DGGE指纹图谱[9]。
1.2.5 乳杆菌属和双歧杆菌属类群定性分析 应用乳杆菌属和双歧杆菌属内常见种的特异性引物(表2)对粪便菌体基因组DNA进行PCR扩增[14-16],定性分析各目标细菌在样品中的出现频率。乳杆菌属(Lactobacillus)检测种包括L.plantarum(植物乳杆菌)、L.salivarius(唾液乳杆菌)、L.casei(干酪乳杆菌)、L.acidophilus(嗜酸乳杆菌)、L.helveticus(瑞士乳杆菌)和L.fermentum(发酵乳杆菌);双歧杆菌属(Bifidobacterium)检测种包括B.longum(长双歧杆菌)、B.animalis(动物双歧杆菌)、B.breve(短双歧杆菌)和B.adolescentis(青春双歧杆菌)。扩增体系(25 μL)为:10×PCR缓冲液 2.5 μL,dNTPs (2.5 mmol/μL)2 μL,引物(10 pmol/μL)各1 μL,模板DNA 1 μL,Taq DNA聚合酶 (5 U/μL)0.25 μL,ddH2O补足至25 μL。反应程序为:95℃ 4 min;95℃1 min,退火30 s,72℃45 s,30个循环;72℃10 min。得到的PCR产物用1 g/dL的琼脂糖凝胶电泳进行检测[14]。
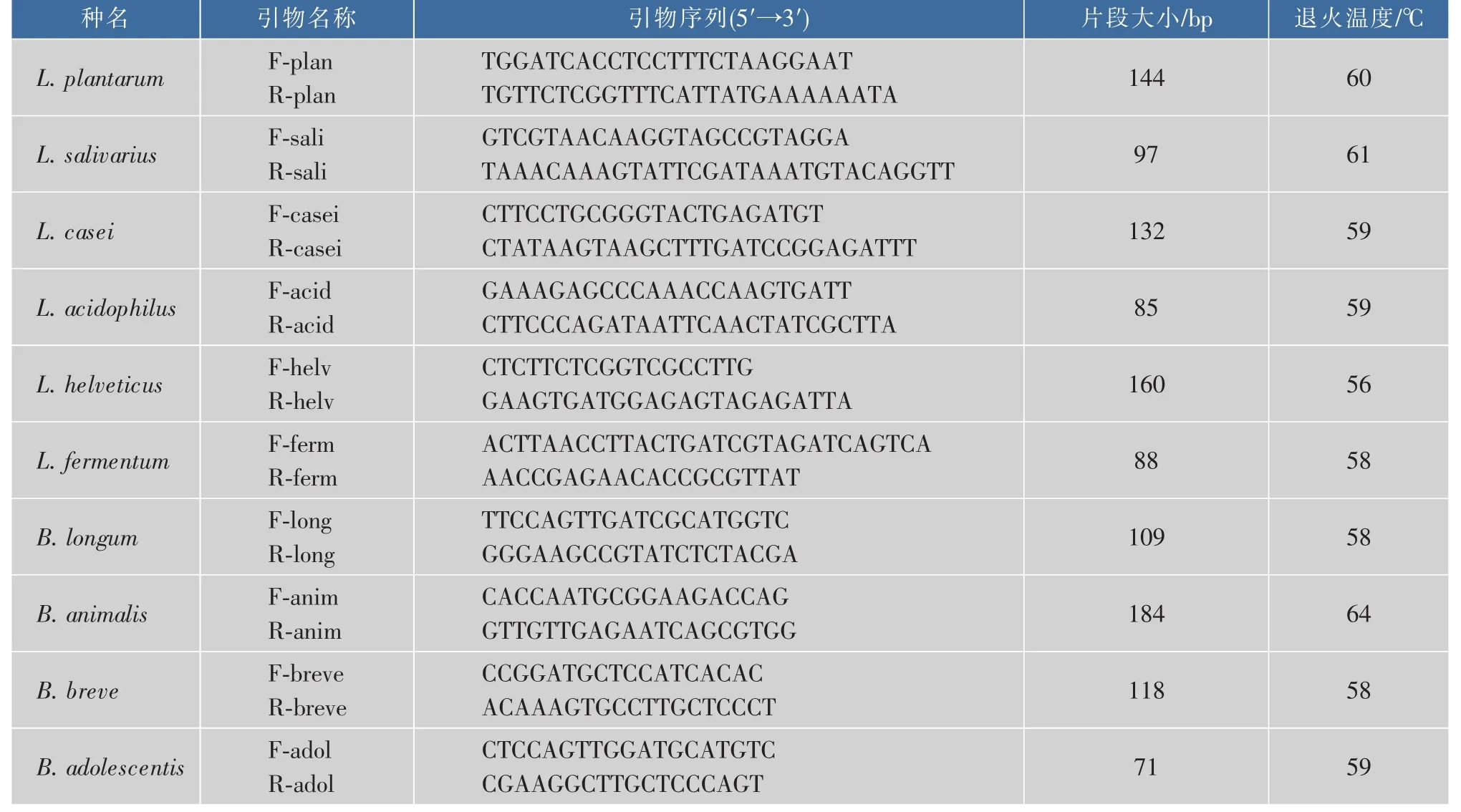
表2 乳杆菌属和双歧杆菌属部分种特异性引物Table2 Specific primers of Lactobacillus and Bifidobacterium Species
1.3 数据处理
1.3.1 统计学分析 基于Past(Hammer等,2001)软件,使用Mann-Whitney法对城乡样本营养成分摄入量和肠道微生物多样性指数进行差异显著性分析。
1.3.2 DGGE图谱聚类分析和PCA分析 使用Quantity One软件对DGGE图谱条带去除背景强度和设定噪声水平,自动获得图谱密度剖面图,完成条带检测,利用条带间相似度采用戴斯系数(Dice coefficient)计算各样品间相似性,得到样品相似性矩阵图,并用UPGMA算法绘制系统树状图,完成图谱聚类分析;同时使用Past软件对相似性矩阵进行PCA分析,并用Origin 7.0(Microcal,USA)绘图软件绘图,得到基于PC1和PC2的赋值样本PCA分布图[11,17]。
1.3.3 样品微生物群落多样性统计学分析 使用多样性指数Shannon-Weiner指数(H)、丰度(S)和均匀度(EH)等指标比较样品的微生物多样性[11,17]。计算公式为

公式(1)(2)(3)中,s为每条泳带中条带数目总和,ni为单一条带i的峰面积,N为所有峰的总面积,Pi为特定条带亮度相对于所有条带总亮度的比率,Hmax是多样性指数最大值。通过软件自动获得图谱密度剖面图并利用条带间相似度按照上述公式进行计算。
1.3.4 DGGE条带的序列测定 选择DGGE图谱中清晰的共性和特异性条带,标记后切胶回收,测序。测序在上海美吉生物医药科技有限公司进行[11]。
2 试验结果
2.1 无锡城乡居民肠道微生物PCR-DGGE指纹图谱
对样本通过基因组DNA提取,16S rRNA V3区PCR扩增和DGGE分析,得到样本PCR-DGGE指纹图谱(图1),每一条泳道代表一位志愿者肠道菌群的DGGE指纹图谱,不同位置的条带代表该样本中不同细菌的16S rDNA V3区基因片段,泳道中条带数量越多,则说明该样本中含有的微生物丰度越高。

图1 城乡居民肠道微生物DGGE指纹图谱Fig.1 DGGE profiles of intestinal microflora for urban and rural residents
由图1可知,各样本含有的条带数目不等、位置各异、亮度不同,说明无锡青年居民肠道微生物结构多样性丰富,且存在个体差异。
2.2 基于PCR-DGGE指纹图谱的城乡青年居民肠道微生物多样性分析
DGGE指纹图谱中,每个泳道中电泳条带的多少直观地反映样本中细菌群落的多样性,条带的亮度反映该细菌的相对生物量,泳道中条带越多,亮度越亮,则表示该样本中微生物多样性越丰富,该种属细菌的相对数量越多。研究中,依据电泳图谱各条带信息,使用Quantity one软件计算各样本丰度(S)、均匀度(EH)和 Shannon-Weine多样性指数(H),以综合分析城乡样本的肠道微生物多样性(表3)。由表3可知,各样品均匀度相似,多样性指数不同,丰度迥异,这个结果与DGGE图谱的直观观测结果一致。城市的12号样本丰度和多样性指数最高,分别为44和3.628 0,表明12号志愿者肠道微生物多样性最高;乡村的7号样本丰度和多样性指数最低,分别为25和3.005 2,表明7号志愿者肠道微生物多样性最低。分别对城乡样本各丰度、多样性指数和均匀度求算术平均值,可知,城市样本的均匀度、丰度和多样性指数均大于乡村样本,分别为0.961 7、36.25和3.426 2,0.942 6、35.50和3.350 5。对结果进行差异显著性分析,均无显著性差异(P>0.05),暗示无锡城乡青年居民肠道微生物多样性差异不显著。
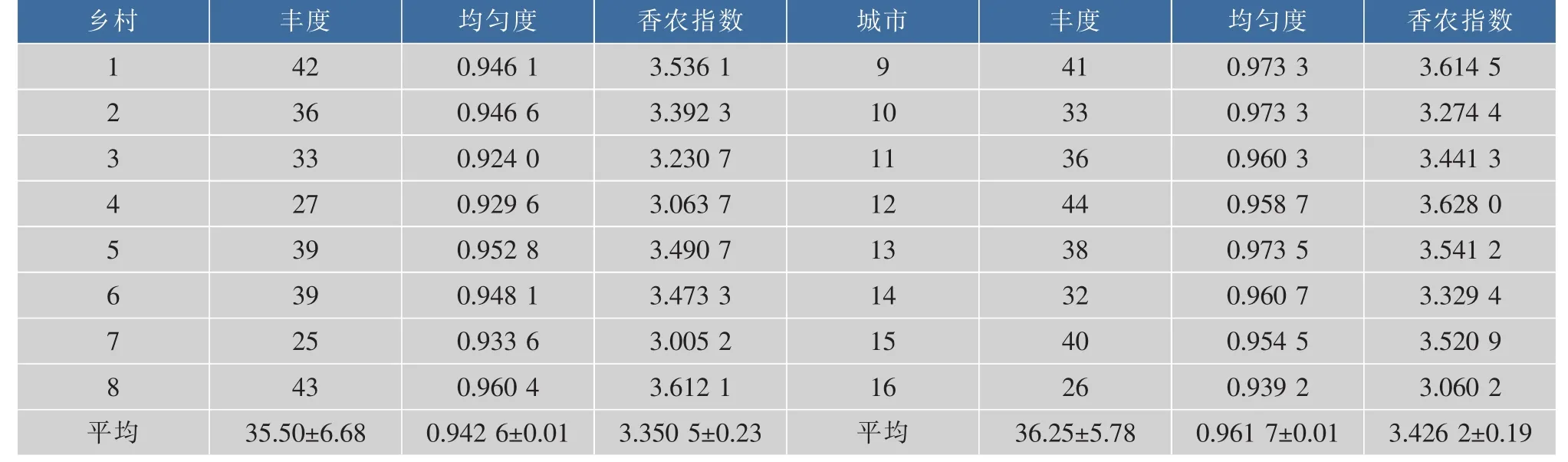
表3 城乡居民样本DGGE条带多样性指数、丰度及均匀度Table3 Shannon-Wiener Index(H),Richness(S)and Evenness(EH)of DGGE bands for urban and rural residents
2.3 基于PCR-DGGE指纹图谱的城乡青年居民肠道微生物相似性分析
基于DGGE指纹图谱,计算各样品间群落相似性系数可以得到各样品细菌种类差异信息。表4为样品间微生物群落相似性分析结果。整体而言,各样品间的微生物群落相似性系数介于 18.5%~72.5%,系数较低,变化区间较大,说明各样本微生物群落结构差异较大,个体差异明显,此结果与DGGE图谱直观观测结果以及多样性分析结果一致。其中,相似性系数较高的有乡村样本的3号和6号、5号和6号,系数分别为71.6%、72.5%;城市样本的9号和12号,14号和15号,系数分别为66.5%、60.9%。相似性系数较低的有乡村的4号和城市的12号,乡村的2号和城市的15号,以及乡村的3号和城市的12号样本,分别为18.5%、25.3% 和22.4%,城市样本和乡村样本内部相似性系数较高,城乡样本间相似性系数较低,说明城市和乡村青年居民内部肠道微生物群落结构相似性较高,大于城乡青年居民之间。此外,表4也显示,部分城乡样本间相似性系数也较高,如乡村的8号和城市的13号样本,乡村的4号和城市的10号样本,系数分别为64.1%、58.4%;而部分城乡样本内部相似性系数较低,如乡村的2号和3号样本,以及城市的11号和12号样本,相似性系数为22.2%和27.7%,说明部分城乡居民间肠道微生物群落结构相似性也较高,存在个体差异。
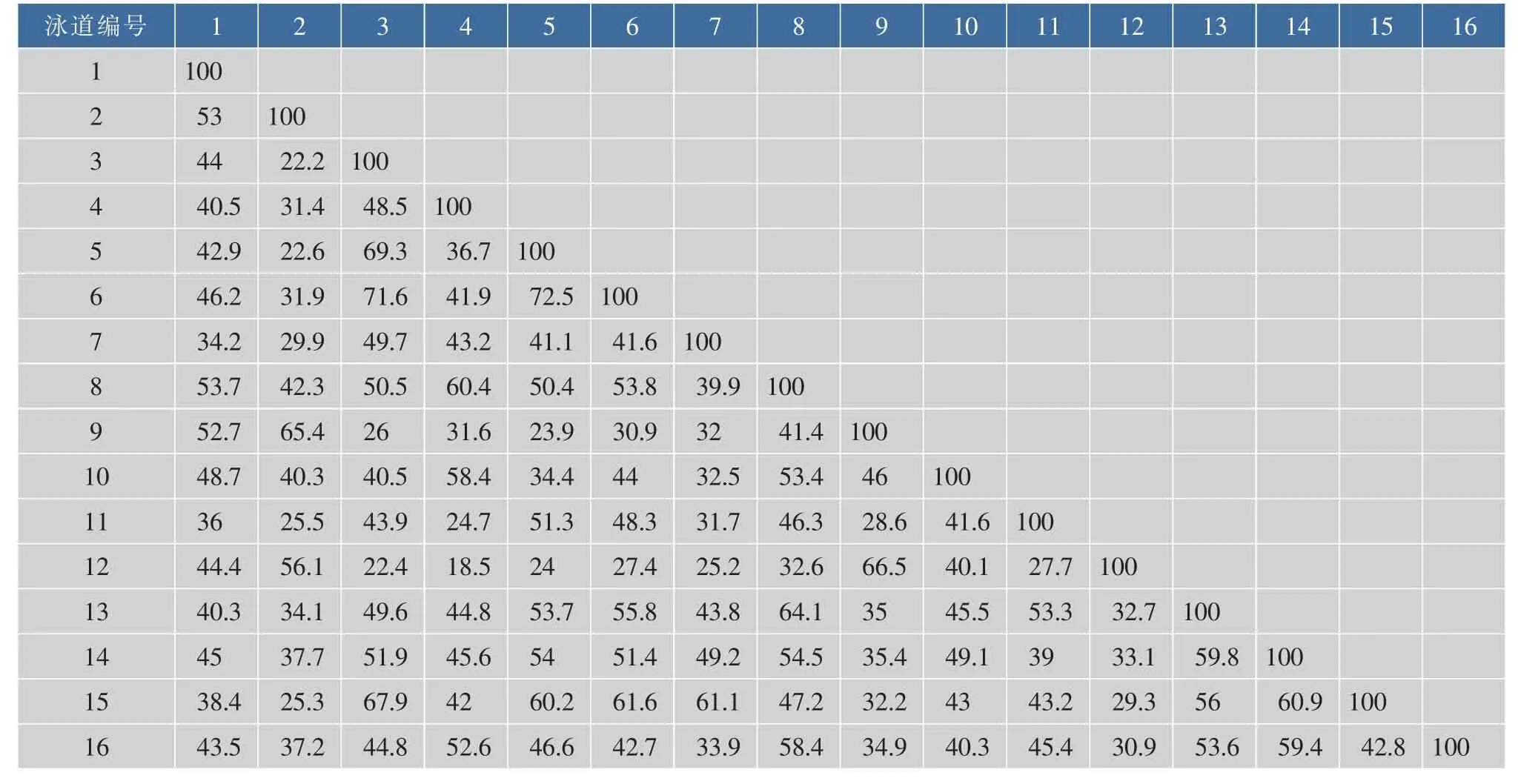
表4 城乡居民肠道微生物群落结构相似性分析Table4 Similarity coefficients of microbial community of urban and rural residents %
对图谱进行聚类分析以进一步观察无锡青年居民肠道微生物群落结构相似性,见图2(a),样本共聚为4簇(族群1—4)。族群1中,城市的9号和12号样本以约66%的相似性聚为一簇;族群2中,城市的14号和15号样本以约60%的相似性聚为一簇;族群3中,乡村的3号、5号和6号样本以约69%的相似性聚为一簇;族群4中,城市的11号、13号和16号样本以约40%的相似性聚为一簇。族群2(城市样本)和族群3(乡村样本)以约35%的相似性聚为一簇,城市或乡村样本按照采集地以较高的相似性聚为一簇,城乡样本间以较低的相似性聚为一簇,说明无锡城市或乡村各自青年居民内部肠道微生物群落结构相似性较高,城乡间则存在差异。族群1、2和4中,乡村的2号、7号、1号和8号样本分别以约63%、53%、40%和80%的相似性与城市样本集聚为一簇,以及族群4中,城市的10号样本和乡村的4号样本以58%的相似性聚为一簇,部分乡村样本以一定的相似性与城市样本集聚,暗示部分乡村青年居民肠道微生物群落结构与城市青年居民相似,这也提示,城乡比较而言,城市居民的肠道微生物群落结构相似性更高,个体差异较小,而乡村居民肠道微生物群落结构个体差异较大。
对图谱进行PCA分析以进一步观察样本间的集聚趋势,如图2(b)所示,主成分因子PC1的贡献率为28.07%,主成分因子PC2的贡献率为16.45%;乡村样本集聚于图形左侧,而大部分城市样本集聚于图形右侧,城乡样本分别集聚为一簇,并彼此分开,说明无锡城乡青年居民肠道微生物群落结构具有分开趋势;此外,也有两个城市样本与乡村样本集聚为一簇的现象存在,说明部分无锡城乡青年居民肠道微生物群落结构相似。
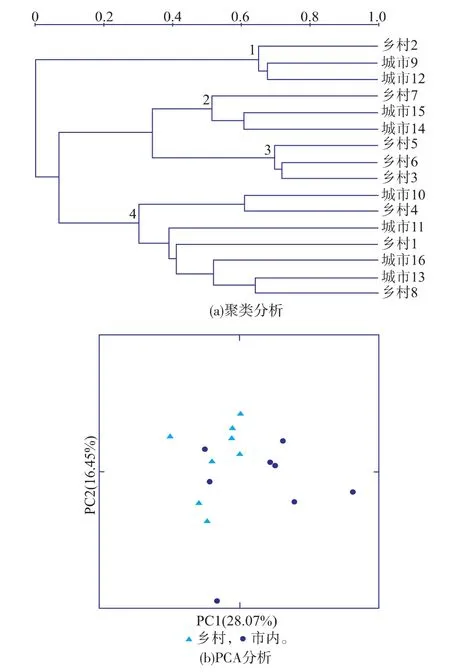
图2 基于DGGE指纹图谱的聚类和PCA分析Fig.2 Analysis of cluster and PCA on DGGE profiles
相似性分析说明,居住距离较远、生活环境不同的无锡城乡青年居民间肠道微生物群落结构差异较小,相似性小于居住距离较近、生活环境相似的无锡城市或乡村居民内部。
2.4 基于PCR-DGGE指纹图谱对城乡青年居民肠道微生物组成的分析
DGGE指纹图谱不仅可直观地反映样本细菌群落结构和多样性,也可进一步通过切胶回收测序分析样本细菌群落组成。研究中,选取图谱中具有代表性的共性和特异性条带(图1)进行胶回收和克隆测序(表5),以分析不同人群肠道菌群组成的异同。试验中共选取条带32条,其中11条条带为所有样本共有,分别为条带1、12—20和25,测序结果显示分别为变形菌门 (Proteobacteria)的不动杆菌属(Acinetobacter),变形菌纲(Alpha proteobacterium)、放 线 菌 门 (Actinobacteria) 的 双 歧 杆 菌 属(Bifidobacterium);条带2—10、21、23—24、26—29 和31—32共18条条带是部分城乡样本共有,测序结果为变形菌门 (Proteobacteria)的不动杆菌属(Acinetobacter)和杆菌属菌,硬壁菌门(Firmicutes)的葡萄球菌属 (Staphylococcus)、 乳 杆菌 属(Lactobacillus)、瘤胃球菌属(Ruminococcus)和梭菌属(Clostridium);条带11、22和30共3条条带为仅乡村样本含有而城市样本不含有,测序结果为杆菌属(Bacterium)菌和变形菌门的不动杆菌属菌。
测序结果中,绝大部分条带的序列与GenBank数据库中细菌的同源性大于98%,有的同源性甚至达到100%。但是条带2、10—13、21、23—24、28和31与数据库中最近亲缘菌种间的同源性低于97%,其鉴定结果分别为不可培养杆菌属细菌、长双歧杆菌和Alpha变形菌纲细菌,因此它们所代表的细菌有可能为新的不可培养物种及其属细菌的新菌种。
测序结果表明,城乡青年居民肠道微生物组成在门水平上相同,都含有硬壁菌门、放线菌门和变形菌门;在属水平上没有多样性的差异,但是丰度和数量上存在差异,其中不动杆菌属、双歧杆菌属、葡萄球菌属、乳杆菌属、梭菌属和瘤胃球菌属为城乡青年居民肠道内的共有菌属。
2.5 乳杆菌属和双歧杆菌属类群定性分析结果
肠道内以乳杆菌属和双歧杆菌属为代表的益生乳酸菌,因其可通过代谢食物中的糖类物质生成乳酸及其它代谢物而保护肠道上皮细胞,加强肠道自身免疫系统功能,抵抗肠道肿瘤及炎症等疾病,这些成了近些年食品微生物学界研究的热点。本课题研究中使用PCR技术对乳杆菌属和双歧杆菌属的若干个种在无锡城乡青年志愿者肠道菌群中的出现频率进行检测(表6)。使用琼脂糖凝胶电泳观察各PCR扩增产物,如果出现清晰、明亮的条带,则扩增成功,说明该样品中含有该目标细菌,否则则说明该样品中不含有该目标细菌。结果显示,乳杆菌属细菌中的L.plantarum、L.casei和L.salivarius以及双歧杆菌属的B.longum和B.breve的检出率较高,为90%左右,在无锡青年居民肠道内普遍存在;乳杆菌属的L.acidophilus和双歧杆菌属的B.animalis和B.adolescentis的检出率适中,约50%~70%,在大部分青年居民肠道内存在;L.helveticus和L.fermentum的检出率较低,为20%左右,在部分城乡青年居民肠道内存在。城乡比较而言,L. plantarum、L.fermentum、L.salivarius、B.animalis、B. breve和B.longum的检出率乡村样本高于城市样本,L.helveticus和L.acidophilus的检出率城市样本高于乡村样本,L.casei和B.adolescentis的检出率城乡样本相同。使用Mann-Whitney分析对各类菌群在城乡样本中的分布进行差异性分析,结果显示均无显著性。
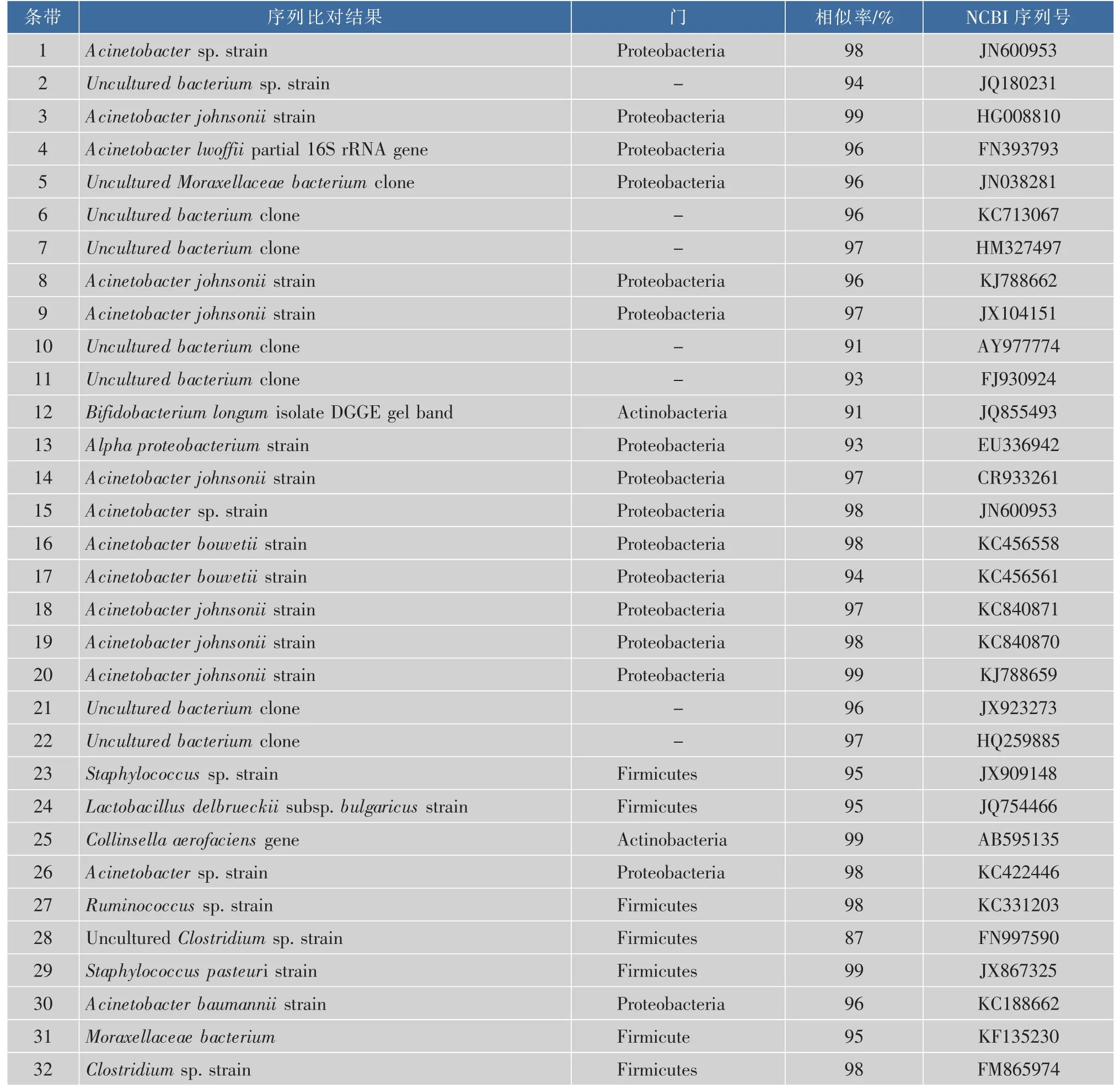
表5 肠道菌群PCR-DGGE特征条带序列比对结果Table5 Alignment of intestinal microflora PCR-DGGE band to its most-similar GenBank sequence
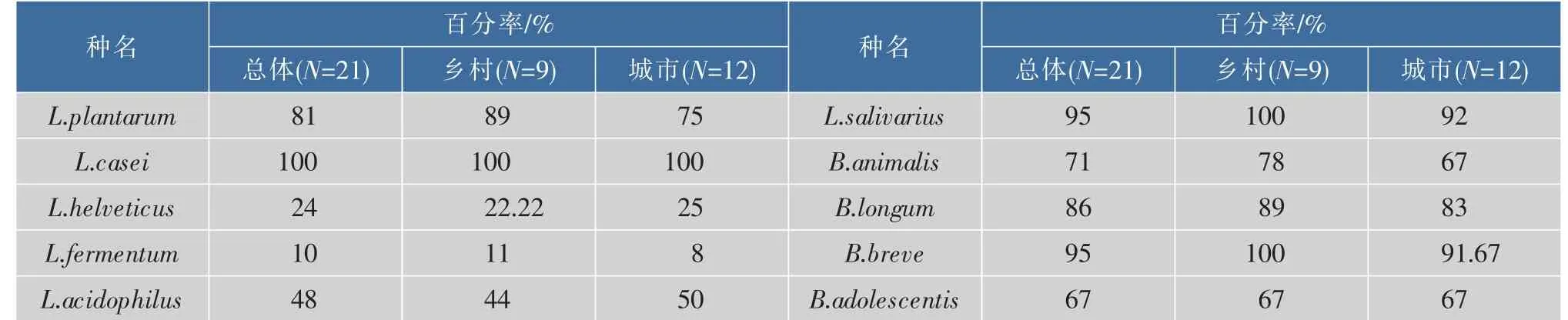
表6 城市和乡村居民肠道中乳杆菌属和双歧杆菌属各种的定性分析Table6 Qualitative analysis of some Lactobacillus and Bifidobacterium species in intestinal tract of urban and rural residents
分析可知,L.plantarum、L.casei、L.salivarius和L.acidophilus以及B.longum、B.breve、B.animalis 和B.adolescentis分别为无锡青年居民肠道内的优势乳杆菌和双歧杆菌。城乡比较而言,各类群菌在居民肠道内分布差异不显著。
3 讨论
个体肠道微生物在经历了婴儿期的不稳定期后,随着个体的成长逐渐成为一个稳定的微生态系统,这个微生态系统受人的基因型、性别、年龄、身体质量指数、地理环境、膳食结构,以及生活方式等因素的影响,不同个体肠道菌群组成不同[1,5]。本课题研究中,DGGE图谱直观观测结果和基于图谱的相似性分析均显示,无锡青年居民肠道微生物群落结构存在个体差异。与本研究结果相似,Zoetendal等[18]在使用TGGE技术对不同个体肠道微生物群落研究的报道中指出,即使是生活在相同环境、具有相同饮食习惯的配偶间,肠道菌群也会显示出较低的相似性。
尽管人体肠道微生物菌群构成个体差异较大,但研究发现不同个体间有相同的核心菌群存在[19-20],而生活环境和饮食方式是引起核心菌群波动的主要因素[7-8]。Yatsunenko等[8]对具有西方饮食习惯的美国居民和非西方饮食习惯的非裔马拉维及土著印第安居民的肠道菌群进行研究,结果发现美国居民与马拉维和印第安居民的肠道微生物群落结构明显分离,集聚为不同的簇。Zhang等[21]在使用PCRDGGE技术对保持原有生活方式和饮食习惯的锡林郭勒盟草原蒙古族居民和已趋于现代化生活方式的呼和浩特城市蒙古族居民的肠道微生物群落结构的研究中证实,城市蒙古族志愿者与草原蒙古族志愿者肠道菌群结构具有显著差异。郭壮[22]亦使用焦磷酸测序技术对西藏藏区居民和城市藏族居民肠道微生物群落结构进行研究,结果显示藏族城市居民样本与藏区居民样本肠道微生物群落结构呈现出明显的分离现象。本研究中得到了与报道相似的结果,基于DGGE图谱进行的相似性分析显示,无锡城市和乡村青年居民肠道微生物群落结构呈现分开趋势,相似性小于城市或乡村青年居民内部。不同的生活环境和生活方式使无锡城乡青年居民肠道微生物群落结构不同。
研究中,基于图谱的多样性分析也显示无锡城乡青年居民肠道微生物多样性差异不显著,定性分析也暗示乳杆菌属和双歧杆菌属各类群细菌在城乡青年居民肠道内分布无显著性差异。与本研究结果不同,Filippo等[23]报道,生活在非洲布基纳法索(Burkina Faso)村庄的儿童其肠道菌群的多样性大于意大利弗罗伦萨(Florence)的城市儿童。郭壮[22]报道,藏族城市居民肠道菌群的多样性高于藏区牧民。近些年随着社会的快速发展,城乡差距逐渐缩小,饮食方式也日趋同化,结合居民膳食调查表进行分析发现,城乡青年居民日常膳食中蛋白质、脂肪、碳水化合物等营养元素的摄入量相当,无显著性差异。这些都足以说明无锡城乡青年居民日常饮食结构已趋相似,相似的饮食习惯可能是引起城乡青年居民肠道微生物多样性差异不显著的原因。此外,本研究中,研究对象分别选自同一省市,距离较近(100 km以内),居民生活环境相似,相似的生活环境可能是引起这一结果的另一原因。
4 结语
综上所述,无锡城市和乡村青年居民肠道微生物多样性差异不显著。通过DGGE技术和PCR技术相结合的方法分析无锡城市和乡村青年居民肠道菌群多样性的异同,以及对优势菌属乳杆菌属和双歧杆菌属各细菌种在志愿者肠道中的分布差异进行初步的分析,揭示了无锡城乡青年居民的肠道微生物多样性,为后续国内人群肠道微生物群落结构的进一步深入研究提供了先例和基础数据。
[1]TURNBAUGH P J,LEY R E,HAMADY M,et al.The human microbiome project[J].Nature,2007,449:804-810.
[2]ECKBURG P B,BIK E M,BERNSTEIN C N,et al.Diversity of the human intestinal microbial flora[J].Science,2005,308:1635-1638.
[3]ZHU B L,WANG X,LI L J.Human gut microbiome:the second genome of human body[J].Cell,2010,1(8):718-725.
[4]MARQUES T M,CRYANA J F,FERGUS S,et al.Gut microbiota modulation and implications for host health:dietary strategies to influence the gut-brain axis[J].Innovative Food Science and Emerging Technologies,2014,22:239-247.
[5]CLEMENTE J C,URSELL L K,PARFREY L W,et al.The impact of the gut microbiota on human health an integrative view[J]. Cell,2012,148:1258-1270.
[6]FINEGOLD S M,ATTEBERY H R,SUTTER V L.Effect of diet on human fecal flora:comparison of Japanese and American diets[J].The American Journal of Clinical Nutrition,1974,27(12):1456-1469.
[7]WU G D,CHEN J,HOFFMANN C,et al.Linking long-term dietary patterns with gut microbial enterotypes[J].Science,2011,334(6052):105-108.
[8]YATSUNENKO T,REY F E,MANARY M J,et al.Human gut microbiome viewed across age and geography[J].Nature,2012,486(14):226.
[9]刘新宇.土壤细菌16S rRNA基因V3区种属特异性检测的法医学应用研究[D].沈阳:中国医科大学法医学院,2008.
[10]YOSHIDA A,SEO Y,SUZUKI S,et al.Actinomycetal community structures in seawater and freshwater examined by DGGE analysis of 16S rRNA gene fragments[J].Marine Biotechnology,2008(10):554-563.
[11]刘慧杰,杨彩云,田蕴,等.基于PCR-DGGE技术的红树林区微生物群落结构[J].微生物学报,2010,50(7):923-930. LIU Huijie,YANG Caiyun,TIAN Yun,et al.Analysis of microbial community structure in mangrove sediments by PCR-DGGE technique[J].Acta Microbiologica Sinica,2010,50(7):923-930.(in Chinese)
[12]杨月欣,王光亚,潘兴昌.中国食物成分表[M].第2版.北京:北京大学医学出版社,2009.
[13]TANAKA S,KOBAYASHI T,SONGJINDA P,et al.Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota[J].FEMS Immunology and Medical Microbiology,2009,56(1):80-87.
[14]徐洁.四川地区不同年龄健康人肠道菌群的比较研究[D].呼和浩特:内蒙古农业大学食品科学与工程学院,2012.
[15]ŠTŠEPETOVA J,SEPP E,KOLK H,et al.Diversity and metabolic impact of intestinal Lactobacillus species in healthy adults and the elderly[J].British Journal of Nutrition,2011,105(8):1235-1244.
[16]JUNICK J,BLAUT M.Quantification of human fecal Bifidobacterium species by use of Quantitative Real-Time PCR analysis targeting the groEL gene[J].Applied and Environmental Microbiology,2012,78(8):2613-2622.
[17]高亦豹,王海燕,徐岩.利用PCR-DGGE未培养技术对中国白酒高温和中温大曲细菌群落结构的分析[J].微生物学通报,2010,37(7):999-1004. GAO Yibao,WANG Haiyan,XU Yan.PCR-DGGE Analysis of the bacterial community of Chinese liquor high and medium temperature Daqu[J].Microbiology China,2010,37(7):999-1004.(in Chinese)
[18]ZOETENDAL E G,AKKERMANS A D,De VOS W M.Temperature gradient gel electrophoresis analysis of 16SrRNA from human fecal samples reveals stable and host-specific communities of active bacteria [J].Applied and Environmental Microbiology,1998,64(10):3854-3859.
[19]ARUMUGAM M,RAES J,PELLETIER E,et al.Enterotypes of the human gut microbiome[J].Nature,2011,473(7346):174-180.
[20]HAMADY M,KNIGHT R.Microbial community profiling for human microbiome projects:Tools,techniques,and challenges[J]. Genome Research,2009,19(7):1141-1152.
[21]ZHANG J,ZHENG Y,GUO Z,et al.The diversity of intestinal microbiota of Mongolians living in Inner Mongolia,China[J]. Beneficial Microbes,2013,4(4):319-328.
[22]郭壮.应用焦磷酸测序技术对不同人群肠道微生物群落结构的研究[D].无锡:江南大学食品学院,2013.
[23]FILIPPO C D,CAVALIERI D,PAOLA M D,et al.Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa[J].Proceedings of the National Academy of Sciences of the United States of America,2010,107(33):14691-14696.
Study on the Diversity of Intestinal Flora of Young Rural and Urban Residents in Wuxi
QIAO Jianmin, ZHANG Jiachao, ZHENG Yi, HOU Qiangchuan,HUANG Weiqiang, HUO Dongxue, GUO Zhuang, ZHANG Heping*
(Key Laboratory of Dairy Biotechnology and Engineering,Ministry of Education,Inner Mongolia Agricultural University,Hohhot 010018,China)
The gut microbial diversity of young people living in rural and urban Wuxi was studied by the combination of polymerase chain reaction(PCR)and denaturing gradient gel electrophoresis (DGGE).Based on the DGGE profile,the similarity of volunteers was performed by analysis of clustering and PCA.On the other hand,the diversity was evaluated by the Shannon-Weine index,richness and eveness.The composition of gut microbiota was further characterized by sequencing of the common and special bands of the lanes in DGGE profile.Based on the PCR,the diversity ofLactobacillus and Bifidobacterium species in the gut of rural and urban volunteers was analyzed.The results showed that the structure of gut microbiota between rural and urban residents of Wuxi appeared a separated tendency,and the similarity of intestinal flora between them was lower than that of urban or rural residents alone.Furthermore,the diversity between them were not significantly different.The genus of Acinetobacter,Bifidobacterium,Staphylococcus,Lactobacillus,Clostridium and Ruminococcus were the common bacteria in the gut of young urban and rural residents.All kinds of bacteria of Lactobacillus(L.)and Bifidobacterium(B.)in the gut were not significantly different in rural and urban residents.And the species of L.plantarum,L.casei,L.salivarius,L.acidophilus,B. longum,B.breve,B.animalis and B.adolescentis were the predominant Lactobacillus and Bifidobacterium bacteria,respectively.On the base of the above comprehensive analysis,it can be preliminarily draw a conclusion that the diversity of gut microbiota was not significantly different between the young residents from rural and urban Wuxi.
young rural and urban residents in Wuxi,microbial diversity,PCR,DGGE
Q 93-3
A
1673—1689(2016)08—0839—10
2014-12-09
农业部现代农业产业技术体系建设项目(CARS-37)。
张和平(1965—),男,内蒙古四子王旗人,农学博士,教授,主要从事乳品生物技术研究。E-mail:hepingdd@vip.sina.com
猜你喜欢
河南医学研究(2022年19期)2022-10-19 00:44:18
数学物理学报(2022年5期)2022-10-09 08:56:44
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
河北画报(2020年8期)2020-10-27 02:54:20
生态学报(2019年11期)2019-07-08 06:18:58
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
水生生物学报(2015年1期)2015-02-28 16:01:05
食品工业科技(2014年23期)2014-03-11 18:19:08
河南科技(2014年18期)2014-02-27 14:14:54
