
不同艾灸因素对阿尔茨海默小鼠PI3K/AKT通路与皮质β淀粉样蛋白沉淀的影响
2016-09-22 05:46刘耀萌刘钧天赵百孝
世界中医药 2016年8期
刘耀萌 刘钧天 黄 畅 韩 丽 赵百孝
(北京中医药大学针灸推拿学院,北京,100029)
不同艾灸因素对阿尔茨海默小鼠PI3K/AKT通路与皮质β淀粉样蛋白沉淀的影响
刘耀萌刘钧天黄畅韩丽赵百孝
(北京中医药大学针灸推拿学院,北京,100029)
目的:观察艾灸、艾烟、无烟灸干预对APP/PS1小鼠大脑皮质PI3K/AKT通路相关指标和β淀粉样蛋白沉淀的影响,从能量代谢的角度探索艾灸不同作用因素在防治阿尔茨海默病中的机制。方法:将32只5个月龄雄性APP/PS1阿茨海默病模型小鼠随机分为模型组、艾灸组、艾烟组、无烟灸组,每组各8只,并将8只同月龄雄性C57BL/6小鼠作为空白对照组。空白组、模型组小鼠每日常规抓取、固定于正常环境;艾灸组与无烟灸组小鼠每日抓取、固定后取关元穴灸治;艾烟组小鼠每日抓取、固定后暴露于10~15 mg/m3艾烟环境。所有干预20 min/d,6 d/周,共8周。于第10周取材。ELISA法检测各组小鼠大脑皮质中IRS-1、PI3K、AKT、GSK-3α、IDE的含量,刚果红染色检测大脑皮质区β淀粉样蛋白沉淀情况。结果:皮质中IRS-1含量比较显示,模型组低于空白组(P<0.01),艾灸组、艾烟组高于模型组(P<0.01,P<0.05);脑中PI3K含量比较显示,模型组低于空白组(P<0.01),艾灸组、艾烟组高于模型组(P<0.05);脑中AKT含量比较显示,模型组低于空白组(P<0.05),艾灸组、艾烟组、无烟灸组高于模型组(P<0.05),艾灸组高于无烟灸组(P<0.05);脑中GSK-3α含量比较显示,模型组高于空白组(P<0.01),艾灸组、艾烟组、无烟灸组低于模型组(P<0.01,P<0.05,P<0.05)。模型组小鼠在皮质内可观察到明显的橘红色Aβ沉积,空白对照组明显少于模型组(P<0.01)。艾烟组、艾烟组、无烟灸组在相同皮质区域中,淀粉样蛋白沉淀均有不同程度的减少。结论:艾灸与艾烟可以调整APP/PS1阿尔茨海默病小鼠的脑部能量代谢,此外艾烟还可以增加对β淀粉蛋白的清除,减缓小鼠皮质内β淀粉蛋白沉淀的产生速度,以延缓AD病理进程。
阿尔茨海默病;艾灸;艾烟;无烟灸;PI3K/AKT通路;胰岛素降解酶
阿尔茨海默病(Alzheimer Disease,AD)是一种起病隐匿的慢性退行性神经病变,多发于65岁以上老年人。患者以短期记忆丧失为发病初期表现,随着病情进一步发展,可观察到学习和记忆功能进行性损害及其他大脑功能受累[1],包括意识错乱、判断能力障碍、语言障碍、焦虑、强迫症和幻觉,甚至出现躁狂或抑郁[2]。据世界卫生组织(WHO)和国际阿尔茨海默病协会(ADI)统计,2015年全球有超过4600万人罹患痴呆,其中亚洲患者比例最大,约有2300万患者,预计2050年,全球痴呆患者数量将高达13150万人次[3]。阿尔茨海默病是引起痴呆的最常见原因,也是现代人的第六大死亡因素,虽然全世界每年对AD进行了大量的研究,但临床上仍然缺乏对抗AD的特效药。
艾灸疗法作为中医学特色疗法,已有数千年历史,对阿尔茨海默病的预防和治疗具有一定效果。根据中医基础理论,艾灸具有温补元气、培肾固本的功效,而老年痴呆病位在脑,患者元气不足,脏腑亏虚,则髓海无以充养。
艾燃烧生成物是艾灸过程中艾绒燃烧产生的烟气,与以往所认为的艾灸最主要的效应因素是温热刺激所不同的是,本课题组在前期研究中通过固相微萃取-气相色谱-质谱联用技术发现艾燃烧过程中可产生26种成分,主要为芳香族化合物、酯类、烷烃或含羟基类化合物,它们也存在诸如改善机体氧化损伤、增强神经元保护功能、调节线粒体功能等生物活性[4-6]。
APP/PS1双转基因小鼠是国际公认的,以Aβ为主要致病因素的阿尔茨海默病动物模型,雄性小鼠在6个月龄时即可观察到大脑皮质、海马区出现老年斑[7]。本实验在前期研究的基础上,将艾灸的不同作用因素分离开来,将温热刺激和艾燃烧生成物的作用分别作为干预因素,以APP/PS1小鼠作为阿尔茨海默病模型,以脑组织中PI3K/AKT通路作为作用靶点,来评估艾灸不同因素对阿尔茨海默病病理进程的影响。
1 材料与方法
1.1实验动物及分组本实验选用5个月龄雄性APP/PS1双转基因小鼠作为阿尔茨海默病模型,小鼠体重为(26.32±2.2)g,以具有相同遗传背景的同月龄雄性野生C57BL/6小鼠作为空白对照组,小鼠体重为(26.62±1.96)g。所有小鼠均为SPF级,购买于北京华阜康生物科技股份有限公司(动物许可证号:SLXK京2009-0007),饲养于北京中医药大学SPF屏障实验室,饲养环境由人工控制,干净卫生,通风良好,每日光照12 h,定期紫外线消毒,保持环境温度为20~25 ℃,相对湿度为50%~65%。所有小鼠单笼饲养,予普通饲料喂养(购买自北京维通利华实验动物有限公司),所有小鼠自由进食饮水。
小鼠适应性喂养1周后,干预8周。将32只APP/PS1小鼠按随机数字表法分为模型组、艾灸组、艾烟组、无烟灸组,共4组,将8只C57BL/6小鼠作为空白对照组。
1.2主要实验材料、仪器及试剂定制普通艾条、无烟艾条:规格为直径0.5 cm×长度20 cm,均购买自河南安阳汉医艾绒有限公司;自制小鼠固定器:呈拱桥状,规格约为150 mm×65 mm,底部由白色树脂片制成,中间留有小孔,可暴露小鼠腹部;拱部由透明有机玻璃制成,四周留有小孔,可供小鼠将头部、四肢、尾部伸出并固定;自制固定器支架:规格为30 cm×12 cm×30 cm,玻璃材质,上层可放置小鼠固定器,下层可放置艾条;自制玻璃缸:规格为80 cm×80 cm×60 cm,玻璃材质,顶部可揭开,中间留有操作孔,直径约5 cm;光散射式数字粉尘测试仪:型号为P5L2C,购买自北京宾达绿创科技有限公司;小鼠胰岛素受体基底1(IRS-1)ELISA实验试剂盒(E03I0403),小鼠磷脂酰肌醇(-3)激酶(PI3K)ELISA实验试剂盒(E03P0185),小鼠蛋白激酶(AKT)ELISA实验试剂盒(E03P0661),小鼠糖原合成酶激酶3α(GSK-3α)ELISA实验试剂盒(E03G0264),小鼠胰岛素降解酶(IDE)ELISA实验试剂盒(E03I0332),均购买自上海蓝基生物科技有限公司。
1.3实验步骤适应性喂养1周后,将32只APP/PS1小鼠按随机数字表法分为模型组、艾灸组、艾烟组、无烟灸组,共4组,将8只C57BL/6小鼠作为空白对照组。参照《实验针灸学》,采取模拟人体解剖部位与骨度分寸法取小鼠关元穴,予以下实验干预。
1.3.1空白暴露将空白组与模型组小鼠分别抓取,放入自制固定器中,使其头部、四肢、尾部固定,关元穴暴露,固定器置于支架上,每日暴露20 min。
1.3.2艾灸、无烟灸干预1)艾灸干预:将小鼠抓取,放入自制固定器中,使其头部、四肢、尾部固定,关元穴暴露,固定器置于支架上,将点燃的艾条置于固定器下层,使其正对小鼠关元穴,适时调整艾条高度,使艾条顶部与小鼠皮肤距离保持为3~4 cm,艾灸过程中将光散射式数字粉尘测试仪置于支架旁,每隔3 min测量1次,使艾烟浓度稳定在10~15 mg/m3,每日干预20 min。
2)无烟灸干预:将小鼠抓取,放入自制固定器中,使其头部、四肢、尾部固定,关元穴暴露,固定器置于支架上,将点燃的无烟艾条置于固定器下层,使其正对小鼠关元穴,适时调整艾条高度,使艾条顶部与小鼠皮肤距离保持为3~4 cm,每日干预20 min。
1.3.3艾烟干预将艾烟组小鼠抓取,放入自制固定器中,使其头部、四肢、尾部固定,关元穴暴露,将固定器放入自制玻璃缸中,将点燃的艾条从玻璃缸盖上操作孔伸入缸内,使艾烟充满玻璃缸后迅速拿出艾条,关闭操作孔。将光散射式数字粉尘测试仪置于玻璃缸内,每隔3 min测量1次,使艾烟浓度稳定在10~15 mg/m3,若低于该浓度则通过操作孔补充艾烟,若高于该浓度,则打开操作孔,使艾烟浓度恢复至范围内,每日干预20 min。
本研究数据均采用SPSS18.0软件进行统计学分析,数据描述采用(±s)或%表示,计数资料采用χ2检验,计量资料采用t检验,P<0.05,差异具有统计学意义。
1.3.4取材将所有小鼠用10%水合氯醛腹腔注射麻醉(3 mL/kg),待完全麻醉后,打开小鼠胸腔,将灌注针插入左心室。随后,缓缓抬起心脏,找到右心房心耳并将其剪开,打开灌流器,慢慢灌注生理盐水30 mL,待生理盐水冲洗干净后,小心开颅剥取小鼠全脑,冰盒上延矢状缝剖为两半,一半留取皮质,另一半整块放入4%多聚甲醛内。
1.3.5标本制作皮质部分按每毫克组织加入5~10 μL裂解液的比例(1∶5~1∶10)加入RIPA裂解液,制成组织匀浆。将匀浆液用低温离心机离心,保持温度4 ℃,转速为12 000 r/min,离心20 min后用移液枪取上清液。
多聚甲醛内的脑组织固定过夜,流水冲洗30 min,用浓度分别为100%、95%、85%、80%的乙醇脱水,二甲苯透明,浸蜡,常规包块。转化切片机切片,制备脑冠状面切片,每片切片厚度约为6 μm。随后取等间隔的组织切片脱腊脱水,予刚果红溶液浸染20 min,用碱性乙醇分化,蒸馏水水洗,95%乙醇溶液冲洗,无水乙醇脱水,二甲苯透明,中性树胶封固。光镜下(×400)选取细胞数较为恒定的CA3区拍片,比较不同组小鼠的Aβ斑变化。
1.4测试指标及方法采用酶联免疫吸附法测定脑组织IRS-1、PI3K、AKT、GSK-3α、IDE活性,采用刚果红染色法测定Aβ淀粉样蛋白沉淀含量。β淀粉样蛋白沉淀予刚果红染色法观察。

2 结果
2.1各组小鼠皮质中IRS-1、PI3K、AKT、GSK-3α含量比较见表1。

表1 小鼠脑组织中的MDA、NO含量、SOD活性比较
注:与模型组比较*P<0.05,**P<0.01;与无烟灸组比较▽P<0.05。
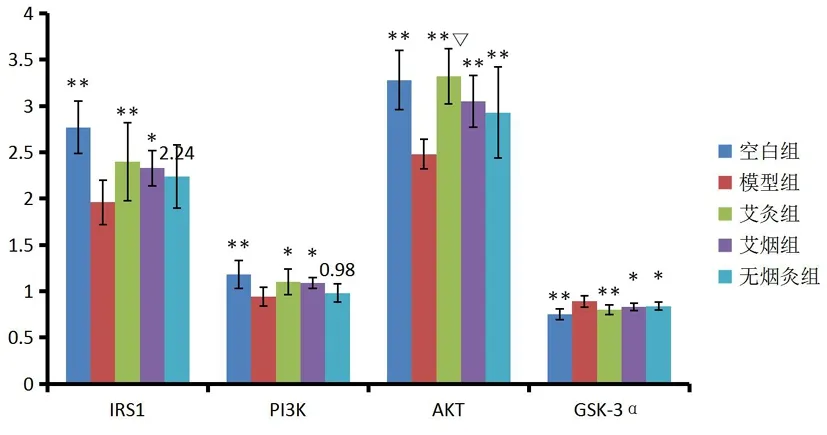
图1 各组小鼠皮质区IRS-1含量
各组小鼠皮质区IRS-1含量(见表1,图1)由高至低分别为:空白组、艾灸组、艾烟组、无烟灸组、模型组。与空白组相比,模型组小鼠大脑皮质IRS-1含量显著降低(P=0.000);与模型组相比,艾灸组、艾烟组小鼠大脑皮质IRS-1含量升高(P=0.008,P=0.017);艾灸组、艾烟组、无烟灸组之间无统计学意义(P<0.05)。
各组小鼠皮质区皮质区PI3K含量由高至低分别为:空白组、艾灸组、艾烟组、无烟灸组、模型组。与空白组相比,模型组小鼠大脑皮质PI3K含量显著降低(P=0.000);与模型组相比,艾灸组、艾烟组小鼠大脑皮质PI3K含量升高(P=0.011,P=0.013);艾灸组、艾烟组、无烟灸组之间无统计学意义(P<0.05)。
各组小鼠皮质区AKT含量由高至低分别为:艾灸组、空白组、艾烟组、无烟灸组、模型组。与空白组相比,模型组小鼠大脑皮质AKT含量显著降低(P=0.000);与模型组相比,艾灸组、艾烟组、无烟灸组小鼠大脑皮质AKT含量升高(P=0.000,P=0.001,P=0.011);与无烟灸组比较,艾灸组AKT含量升高(P=0.032)。
各组小鼠皮质区GSK-3α含量由低至高分别为:空白组、艾灸组、艾烟组、无烟灸组、模型组。与空白组相比,模型组小鼠大脑皮质GSK-3α含量显著升高(P=0.000);与模型组相比,艾灸组、艾烟组、无烟灸组小鼠大脑皮质GSK-3α含量显著降低(P=0.002,P=0.018,P=0.048);艾灸组、艾烟组、无烟灸组之间无统计学意义(P<0.05)。
2.2各组小鼠皮质中IDE含量比较见表2。

表2 小鼠脑组织中的IDE含量比较
注:与模型组比较*P<0.05,**P<0.01;与无烟灸组比较▽P<0.05。
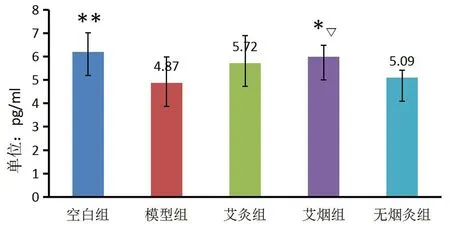
图2 各组小鼠皮质区IDE含量

图3 空白组皮质中β淀粉样蛋白沉淀(×20) 图4 模型组皮质中β淀粉样蛋白沉淀(×20)

图5 艾灸组皮质中β淀粉样蛋白沉淀(×20) 图6 艾烟组皮质中β淀粉样蛋白沉淀(×20)
各组小鼠皮质区IDE含量(见表2,图2)由高至低分别为:空白组、艾烟组、艾灸组、无烟灸组、模型组。与空白组相比,模型组小鼠大脑皮质IDE含量显著降低(P=0.003);与模型组相比,艾烟组小鼠大脑皮质IDE含量降低(P=0.012);与无烟灸组比较,艾烟组IDE含量升高(P=0.048)。
2.3各组小鼠海马Aβ沉淀比较经刚果红染色的APP/PS1小鼠海马皮质区放大20倍在光学显微镜下观察,空白组小鼠,见图3。皮质区未见明显橘红色染色的物质沉积,细胞排列整齐;模型组小鼠,见图4。皮质区可见明显橘红色Aβ沉积(如箭头所指),呈点斑片状,大小不一,细胞排列较散乱、稀疏;艾灸组小鼠,见图5。皮质区可见少量橘红色Aβ沉积(如箭头所指),呈点斑片状,大小不一,细胞排列较整齐;艾烟组小鼠,见图6。皮质区可见少量橘红色Aβ沉积(如箭头所指),呈点斑片状,大小不一,细胞排列较整齐;无烟灸组小鼠,见图7。皮质区可见少量橘红色Aβ沉积(箭头所指),呈点斑片状,大小不一,细胞排列较整齐。
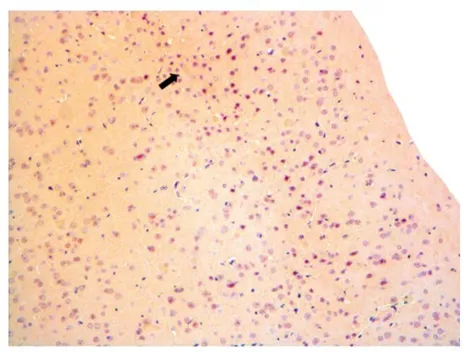
图7 无烟灸组皮质中β淀粉样蛋白沉淀(×20)表3 各组小鼠脑组织海马中淀粉样 蛋白阳性面积比较(μm2)

组别鼠数淀粉样蛋白阳性面积(μm2)空白组8877.79±132.59**模型组813640.42±2756.22艾灸组73228.12±578.99**艾烟组83660.92±846.34**无烟灸组73446.23±237.18**
注:与模型组比较**P<0.01。

图8 各组小鼠脑组织海马中淀粉样 蛋白阳性面积比较(μm2)
各组小鼠皮质中Aβ淀粉样沉淀面积(见表3,图8)由大到小分别为:模型组、艾烟组、无烟灸组、艾灸组、空白组。与空白组相比,模型组小鼠大脑皮质β淀粉样蛋白沉淀阳性面积显著增大(P=0.000);空白组、艾灸组、艾烟组、无烟灸组小鼠均较模型组小鼠阳性面积有显著减少(P=0.000,P=0.000,P=0.000,P=0.000);艾灸组、艾烟组、无烟灸组之间无统计学意义。
3 讨论
磷脂酰肌醇3激酶/蛋白激酶B(Phosphatidylinositol-3-kinase/AKT,PI3K/AKT)信号通路是人体最重要的信号通路之一,它在人体起着承上启下的作用,人体血糖浓度的调节和胰岛素的释放都需要通过该通路完成[8-9]。向上游看,PI3K/AKT信号通路接受脑内胰岛素信号的传导,并发挥重要作用。外周胰岛素通过血脑屏障进入大脑后,人体自身中枢神经系统的神经元也可以在脑内合成胰岛素,释放和在释放神经递质,参与长时程记忆的巩固,同时也在神经元的分化,神经细胞的生长、增殖和修复中发挥着重要作用[10]。当胰岛素受体减少,或胰岛素受体对胰岛素失敏时,可发生类似于阿尔茨海默病的轻型记忆功能减退。同时,临床研究发现,高达60%的AD患者伴随有与2型糖尿病密切相关的医学症状,例如高血糖、胰岛素抵抗等等,均可表明阿尔茨海默病与PI3K/AKT通路有密切的联系[11]。
胰岛素受体底物-1(Insulin Receptor Stubstrate-1,IRS-1)蛋白是调节脑内胰岛素信号转导的最上游的信号转导蛋白,胰岛素与胰岛素受体结合时可使其激活,并发生自身磷酸化,酪氨酸磷酸化的IRS转化细胞调节生长、代谢下游PI3K,刺激葡萄糖转换[12]。IRS-1蛋白在脑组织结构和功能维护起关键性作用,IRS-1基因敲除小鼠会表现出体积较小,尤其是脑质量显著减低的特点[13]。IRS-1的含量减少,表明维持神经元存活和代谢的有效激活下游通路的功能障碍。
IRS激活后的下游信号转导通路,则主要由PI3K途径来完成。P13K是一种胞内磷脂酰肌醇激酶,AKT也是一个重要的蛋白激酶,是PI3K/AKT信号通路中最重要的下游靶目标,具有抗调亡的作用[14-15]。有研究报道,只有激活AKT的表达才能维持细胞的生存。AKT可以通过激活下游底物蛋白,促使其磷酸化,保护神经元,以发挥其抗凋亡的作用;同时,AKT可以直接起到保护线粒体的功能,通过保护完整的膜电位,来达到抑制调亡的目的[16]。
PI3K/AKT信号通路可调节糖原合酶激酶-3(Glycogen Synthase Kinase-3,GSK-3)的活性。GSK-3是一种多功能的丝氨酸/苏氨酸蛋白激酶,在中枢神经系统中起着非常重要的作用[17-19]。GSK-3在哺乳动物体内主要分为GSK-3α和GSK-3β 2种亚型,其中GSK-3α参与调节γ分泌酶,通过针对γ分泌酶特异性底物或接近其复合物的底物来调节γ分泌酶的活性,促使Aβ生成。Ryan等也证实了GSK-3α可以通过增加细胞内成熟的APP,导致AD发病[20];被激活的GSK-3β则可作用于Tau蛋白,使其过度磷酸化并导致神经纤维缠结[21-23],这两方面均能加快阿尔茨海默病的进程。因此当PI3K/AKT信号通路被上调时,AKT激酶会发生磷酸化,AKT的活性被激活,能起到抑制GSK-3α的活性的作用,从这个方向起到抑制Aβ淀粉沉淀生成、阻止细胞调亡的作用。以上过程共同决定了β淀粉样蛋白通过PI3K/AKT通路生成的情况。
同时,正常情况下,脑内Aβ沉淀的水平既取决于脑内Aβ的生成程度,也取决于其清除程度,Aβ可由多种细胞产生,循环于血液、脑脊液和脑间质液中,大部分与侣伴蛋白分子结合,少数以游离状态存在。脑内Aβ清除有多种渠道,Aβ从神经元产生后,可通过酶降解、细胞吞噬隔离和向脑脊液(CSF)、外周血液转运等途径进行清除,当患者罹患阿尔茨海默病时,Aβ淀粉沉淀的生成消除平衡被打破,因此代谢处于失衡状态,进一步加重阿尔茨海默病的进程。
胰岛素降解酶(Insulin Degrading Enzyme,IDE)则为Aβ清除机制中重要的一种Aβ降解酶,与能量代谢密切相关[24-25]。IDE在人体内广泛表达,在代谢较快的肝脏、肾脏、脑组织、肌肉和睾丸组织中表达最为丰富。研究发现IDE可以降解多种小分子多肽,如胰岛素、胰高血糖素、转化生长因子、P-内啡肽等[26-27]。脑内神经元和小胶质细胞可以分泌的IDE,降解Aβ[28],通过对IDE基因敲除小鼠Aβ含量的测量发现,不能正常合成IDE则会造成Aβ降解的减少,其减少程度高达50%[29-30]。胰岛素和Aβ同时属于IDE反应的底物,但胰岛素对IDE的亲和力要远远大于Aβ对IDE的亲和力,因此当机体产生胰岛素抵抗,胰岛素代偿性分泌含量升高时,过量的胰岛素会竞争性的与IDE结合,抑制IDE与Aβ的结合,间接抑制了IDE对Aβ的降解,促进了Aβ的聚集。
本实验模型组中,8个月龄的APP/PS1小鼠,相较同月龄的C57BL/6小鼠,皮质中IRS-1含量、PI3K含量、AKT含量均下降,GSK-3α含量上升,且差异有统计学意义,说明APP/PS1小鼠脑内PI3K/AKT通路已造成损伤。艾灸、艾烟组可使IRS-1、PI3K含量上升,艾灸、艾烟、无烟灸组可使AKT含量上升、GSK-3α含量下降,说明艾灸、艾烟、无烟灸均可调整PI3K/AKT通路,抑制皮质Aβ生成,且三者作用强度无统计学意义。8月龄的APP/PS1小鼠,相较同月龄的C57BL/6小鼠,皮质中IDE含量下降,且差异有统计学意义,说明APP/PS1小鼠脑内IDE介导的Aβ清除机制已造成损伤。艾烟组可使IDE含量上升,且差异有统计学意义,艾灸、无烟灸组也表现出升高IDE含量的趋势,说明艾灸、艾烟、无烟灸均可通过加强Aβ淀粉蛋白清除程度,来抑制皮质Aβ生成,且艾烟组表现出的改善作用最强。
[1]Querfurth HW,LaFerla FM.Alzheimer′s disease[J].N Engl J Med,2010,362(4):329-344.
[2]Burns A,Iliffe S.Alzheimer′s disease[J].BMJ,2009,April.doi:10.1136/bmj.b1349.
[3]Prince M,Albanese E,Guerchet M,et al.World Alzheimer Report 2014-Dementia and Risk Reduction:An analysis of protective and modifiable factors; Alzhermer′s Disease International(ADI):London,UK,2014.
[4]靳然,赵百孝,于密密,等.艾燃烧生成物组分固相微萃取气相色谱质谱法定性分析[J].北京中医药大学学报,2011,34(9):632-636.
[5]靳然,于密密,赵百孝,等.气相色谱法测定艾叶4个挥发性成分的含量[J].药物分析杂志,2013,33(6):1033-1036.
[6]许焕芳,赵百孝.艾灸疗法作用机理浅述[J].上海针灸杂志,2012,31(1):6-9.
[7]朱斌,陈静,秦红芳,等.APP/PS1双转基因老年性痴呆小鼠早期病理和认知行为变化[J].广州中医药大学学报,2012,29(2):193-196,226.
[8]Monte D S M D L.Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer′s disease[J].Drugs,2012,72(1):49-66.
[9]Muta,Kenjiro,Morgan,et al.p110alpha subunit of PI3K is a crucial component of brain insulin receptor signaling in regulating lumbar sympathetic nerve traffic[J].Jurnalul De Chirurgie,2009,106(11):965-970.
[10]Sm D L M.Insulin resistance and Alzheimer′s disease[J].Bmb Reports,2009,42(8):475-481.
[11]Infantegarcia C,Ramosrodriguez J J,Galindogonzalez L,et al.Long-term central pathology and cognitive impairment are exacerbated in a mixed model of Alzheimer′s disease and type 2 diabetes[J].Psychoneuroendocrinology,2015,65:15-25.
[12]Niswender K D,Stearns W H,Meyers M G,et al.Evidence for IRS-PI3K signaling in the hypothalamic response to insulin and leptin[J].Diabetes,2001,50:A13-A13.
[13]Araki E,Lipes M A,Patti M E,et al.Alternative pathway of insulin signalling in mice with targeted,disruption of the IRS-1 gene[J].Nature,1994,372(6502):186-190.
[14]Moloney A M,Griffin R J,Timmons S,et al.Defects in IGF-1 receptor,insulin receptor and IRS-1/2 in Alzheimer′s disease indicate possible resistance to IGF-1 and insulin signalling[J].Neurobiology of Aging,2010,31(2):224-243.
[15]Steen E,Terry B M,Rivera E J,et al.Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer′s disease——is this type 3 diabetes?[J].Journal of Alzheimers Disease Jad,2005,7:63-80.
[16]Bijur G N,Jope R S.Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation[J].Journal of Neurochemistry,2003,87(6):1427-1435.
[17]Sofola O,Kerr F,Rogers I,et al.Inhibition of GSK-3 Ameliorates Aβ Pathology in an Adult-Onset,Drosophila,Model of Alzheimer′s Disease[J].Plos Genetics,2010,6(9):e1001087.
[18]Noh M,Koh S Y,Kim H,et al.Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-beta-induced neuronal cell death[J].Journal of Neurochemistry,2009,108(5):1116-1125.[19]Pena-Ortega F,Solis-Cisneros A,Ordaz B,et al.Amyloid beta 1-42 inhibits entorhinal cortex activity in the beta-gamma range:role of GSK-3[J].Current Alzheimer Research,2012,9(7):857-863.
[20]Ryan K A,Pimplikar S W.Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain[J].Journal of Cell Biology,2005,171(2):327-335.
[21]Xie H Q,Litersky J M,Hartigan J A,et al.The interrelationship between selective tau phosphorylation and microtubule association[J].Brain Research,1998,798(1-2):173-183.
[22]Wang J Z,Zhang Y.Configuration-specific immunotherapy targeting cis,pThr231-Pro232 tau for Alzheimer disease[J].Journal of the Neurological Sciences,2015,348(s 1-2):253-255.
[23]Sivaprakasam P,Han X,Civiello R L,et al.Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core[J].Bioorganic & Medicinal Chemistry Letters,2015,25(9):1856-1863.
[24]Pérez A,Morelli L,Cresto J C,et al.Degradation of Soluble Amyloid β-Peptides 1-40,1-42,and the Dutch Variant 1-40Q by Insulin Degrading Enzyme from Alzheimer Disease and Control Brains[J].Neurochemical Research,2000,25(2):247-255.
[25]Vekrellis K,Ye Z,Qiu W Q,et al.Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme[J].Journal of Neuroscience the Official Journal of the Society for Neuroscience,2000,20(5):1657-1665.
[26]Shen Y,Joachimiak A,Rosner M R,et al.Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism[J].Nature,2006,443(7113):870-874.
[27]Wang D S,Dickson D W,Malter J S.β-Amyloid Degradation and Alzheimer′s Disease[J].Biomed Research International,2006(3):58406.
[28]Leissring M A.Enhanced Proteolysis of β-Amyloid in APP Transgenic Mice Prevents Plaque Formation,Secondary Pathology,and Premature Death[J].Neuron,2003,40(6):1087-1093.
[29]Qiu W Q,Walsh D M,Ye Z,et al.Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation[J].Journal of Biological Chemistry,1998,273(49):32730-32738.
[30]Farris W,Mansourian S,Yang C,et al.Insulin-degrading enzyme regulates the levels of insulin,amyloid β-protein,and the β-amyloid precursor protein intracellular domain in vivo[J].Proc Natl Acad Sci U S A,2003(7):4162-4167.
(2016-06-28收稿责任编辑:洪志强)
Effect of Different Factors of Moxibustion on PI3K/AKT Signal Pathway and Cortical β-amyloid Protein Precipitation in Brain Tissue of Alzheimer Mice
Liu Yaomeng, Liu Juntian, Huang Chang, Han Li, Zhao Baixiao
(BeijingUniversityofChineseMedicine,SchoolofAcupuncture-MoxibustionandTuina,Beijing100029,China)
Objective:To observe the effect of smoking moxibustion, none-smoking/ heat moxibustion and moxa smoke on PI3K/AKT signal pathway and cortical β-amyloid protein precipitation and discuss the mechanism of them in preventing and treating Alzheimer. Methods: Eight wide-type C57BL/6 normal mice served as normal control group, and 32 APP/PS1 Alzhermer model mice were randomly divided into four groups (n=8/group): 1) No-treatment control group, 2) Moxibustion group, 3) Moxa smoke group, 4) one-smoking moxibustion/heat group. The treatment were given 20 mins a day, 6 days a week for 8 weeks. In group 2 and 3, the moxa stick was pointed above acupoint Guanyuan(CV4). In group 3, mice were exposed to moxa smoke (10~15 mg/m3). However in group 1 and normal control group, no treatment was given. ELISA technique was used to detect the level of IRS-1, PI3K, AKT, GSK-3α, and IDE. In addition, β-amyloid was evaluated using Congo red staining.Results: Compared to group 1, mice in group 2, group 3 and group 4 had significant higher levels of IRS-1, PI3K, AKT and lower levels of GSK-3α (P<0.05). Comparing with group 1, mice in group 4 had significant higher levels of IDE (P<0.05). Mice in group 1 expressed great aggregation of β-amyloid while mice in normal control group had none aggregation of β-amyloid (P<0.05). Mice in group 2, group 3, group 4 had less aggregation of β-amyloid (P<0.05). Conclusion: Moxibustion, moxa smoke and heat-treatment moxibustion play equal role of delaying the pathological process Alzheimer′s disease by PI3K/AKT pathway and can induce less aggregation of β-amyloid.
Alzheimer′s disease; Moxibustion; Moxa smoke; Heat-treatment moxibustion; PI3K/AKT pathway; IDE
国家自然科学基金项目(编号:81373730);国家青年自然基金项目(编号:81403449);国家自然科学基金项目(编号:81574068);国家国际科技合作专项项目(编号:2011DFA31370)
刘耀萌(1990—),女,2013级硕士研究生
赵百孝(1963.03—),博士,教授,博士研究生导师,北京中医药大学针灸推拿学院院长,研究方向:艾灸作用与原理,Tel:(010)64286737,E-mail:baixiao100@vip.sina.com
R245.81
A
10.3969/j.issn.1673-7202.2016.08.002
猜你喜欢
中老年保健(2022年2期)2022-08-24
中老年保健(2022年1期)2022-08-17
基层中医药(2021年7期)2021-11-02
成都医学院学报(2021年2期)2021-07-19
中国民间疗法(2021年4期)2021-06-09
世界科学技术-中医药现代化(2021年10期)2021-03-02
小资CHIC!ELEGANCE(2021年46期)2021-01-11
睿士(2020年11期)2020-11-16
基层中医药(2018年7期)2018-12-06
奥秘(2018年9期)2018-09-25
