
抗结核药物致肝毒性易感性研究进展
2016-09-16 05:58:51李颖佳申阿东
中国循证儿科杂志 2016年4期
李颖佳 綦 辉 申 晨 申阿东
·综述·
抗结核药物致肝毒性易感性研究进展
李颖佳綦辉申晨申阿东
近年来,随着结核分枝杆菌耐药菌株的出现和获得性免疫缺陷综合征(AIDS)患者的增加,结核病(TB)成为全球公共卫生日益严峻的挑战和负担。抗结核化疗方案仍以异烟肼(INH)、利福平(RMP)、吡嗪酰胺(PZA)等联合用药为主,然而抗结核药物在治疗过程中引起药物不良反应(ADR),影响了抗结核治疗效果和患者健康。常见的抗结核ADR有抗结核药物致肝毒性(ATDH)、皮疹、胃肠功能障碍和神经系统病变等,其中以ATDH最严重[1],常导致抗结核治疗中断,甚至治疗失败[2]。提高对ATDH的认识,预测并预防其发生和进展有重要临床意义。
1 ATDH概述
已有的资料显示,ATDH发生率5%~28% ,不同地区不同人群中ATDH发生率存在较大差异[3],可能与研究人群不同和对ATDH的定义不同有关[2, 4]。通常对ATDH的定义为:抗结核药物治疗初期突然出现的肝损伤,表现为ALT高于正常值上限(ULN)的2~5倍,伴或不伴有肝脏毒性症状[2, 4]。
ATDH潜伏期为抗结核药物治疗后1周至12个月,中位数为8周[5]。临床表现可不典型,常表现为厌食、恶心、呕吐、食欲不振、上腹部不适、皮肤黄染和茶色尿等非特异性临床症状或体征[5]。肝脏病理呈肝小叶区域性、亚大块性或大块性坏死[5]。临床上ATDH的血生化和组织学特征很难与病毒性肝炎区分[6]。严重肝损伤发生前停止抗结核用药,肝脏功能即可恢复正常[5]。
2 ATDH发病机制
ATDH发病机制较复杂,往往是多种机制先后或共同作用的结果。目前研究认为,引起ATDH的物质主要是抗结核药物的活性代谢产物,如肼、乙酰肼[2]。Ⅰ相代谢酶主要参与药物的生物活化(如氧化、还原或水解),如细胞色素P450(CYP450)酶[6]。药物或其Ⅰ相代谢产物在Ⅱ相代谢酶作用下,与葡萄糖醛酸、硫酸、甘氨酸和谷胱甘肽(GSH)等结合后水溶性增加,利于从体内排出[6];参与Ⅱ相代谢反应的酶主要是谷胱甘肽S转移酶(GST)和尿苷二磷酸葡萄糖醛酸转移酶(UGT)等。Ⅲ相代谢酶如溶质载体(SLC)、三磷酸腺苷结合盒(ABC)[7]等,主要参与药物或其代谢产物的吸收和排泄,也被称为转运体。各相代谢酶活性发生改变时,均有可能造成药物或其代谢产物在体内积累[3]。
一线抗结核药物中,INH、RMP及PZA常引起ATDH[5]。INH主要通过其代谢产物肼、乙酰肼等引起ATDH[4, 6],其代谢途径包括主路途径和旁路途径。如图1所示,在主路途径中,INH被N乙酰基转移酶2(NAT2)乙酰化为乙酰异烟肼,后者被酰胺酶水解成乙酰烟肼后,再继续乙酰化为无毒的二乙酰肼[3, 6];INH也可通过旁路途径直接水解为肼,后者再被NAT2乙酰化为乙酰烟肼[3, 6]。肼具有较强肝毒性,可以引起细胞不可逆性损伤[8]。NAT2乙酰化阶段生成的乙酰烟肼经细胞色素P450 2E1(CYP2E1)催化后,转化为具有较强肝毒性的中间代谢产物,如乙酰偶氮、烯酮和乙酰氧离子等[3]。这些毒性中间代谢产物必须经GST催化,与GSH共价结合并排出体外,

图1异烟肼的代谢过程[8]
注NAT2: N乙酰基转移酶2
从而起到解毒作用[3]。目前,RMP或PZA发生ATDH的具体机制仍不清楚[2]。与INH相似,RMP引起的肝损伤为肝细胞型[5]。RMP单用时可干扰胆红素的吸收和排泄,引起不伴血清转氨酶升高的(直接或间接)高胆红素血症[2, 5];与INH合用时,RMP可以诱导酰胺酶和CYP2E1酶活性,使INH释放的肝毒性物质(如肼)增加,从而增加INH所致肝毒性风险[2, 5]。目前的研究认为,PZA导致的肝毒性具有剂量依赖性[5]。与RMP相似,PZA与INH合用时也能增加患者ATDH发生风险[2]。
活性代谢产物可通过耗竭抗氧化物质或与细胞酶类、脂质或核酸选择性结合,从而引起细胞应激反应[3],线粒体是ATDH发生的重要靶点[3]:细胞应激抑制线粒体呼吸链,导致ATP衰竭和活性氧(ROS)积累。过量ROS积累可以导致脂质过氧化和细胞死亡[3]。红系衍生的核因子2相关因子2(Nrf2)是抗氧化反应的核心转录因子[2],Nrf2-抗氧化反应元件(Nrf2-ARE)信号通路中,抗氧化激活途径和抑制途径的失调在ATDH发生中也发挥了重要作用[9],其具体机制见本文3.3部分。锰超氧化物歧化酶(MnSOD)对线粒体基质中超氧阴离子的解毒具有关键作用[3]。MnSOD功能发生改变时,对超氧化物的解毒缺乏,导致线粒体抑制,最终线粒体通透性发生改变,随之发生ATP依赖性细胞凋亡或坏死[3]。
肝细胞功能障碍和损伤时可激活免疫反应,包括先天性免疫反应和获得性免疫反应[2]。肝损伤时,机体分泌的肿瘤坏死因子-α(TNF-α)、γ干扰素(IFN-γ)、白细胞介素4(IL-4)、IL-6等促炎-抗炎因子调节炎症反应,决定肝细胞发生恢复反应还是损伤反应[2, 3]。至于获得性免疫反应,目前研究者提出2种理论,即半抗原理论和药物与免疫受体的药理作用(p-I)理论[2]。半抗原理论认为,药物或活性代谢产物与肝内蛋白(如CYP酶类)形成的共价复合物(即半抗原),可以诱导抗体或毒性T细胞反应,造成细胞损伤[2, 10]。p-I理论认为某些药物能够模拟配体的作用,可以不与肝内蛋白结合,直接与T细胞受体结合,并以主要组织相容性复合物(MHC)依赖型经典模式使T细胞活化[2, 10]。
3 基因多态性与ATDH易感性
从ATDH发病机制可知,药物代谢酶(DME,Ⅰ相、Ⅱ相)、药物转运体(Ⅲ相代谢酶)、抗氧化反应和免疫反应在ATDH发生发展过程中均起着重要作用,参与这些代谢过程的相关基因的多态性与ATDH易感性密切相关。
3.1DME与ATDH易感性相关的Ⅰ相代谢酶主要有CYP2E1、CYP3A4、CYP2B6和羧酸酯酶1(CES1),Ⅱ相代谢酶主要有NAT2、GST、UGT和MnSOD。
3.1.1Ⅰ相代谢酶
3.1.1.1CYP2E1其活性受CYP2E1基因上多个位点多态性的调控[8]。位于CYP2E1转录起始点上游,分别被限制性内切酶RsaⅠ和PstⅠ识别的2个多态性位点,表现出完全连锁不平衡[2]。野生型等位基因RsaⅠ(+)/PstⅠ(-)被定义为′c1′,命名为CYP2E1*1A;其中任意一个位点发生突变,即RsaⅠ(-)或PstⅠ(+)被定义为′c2′,命名为CYP2E1*5[8]。CYP2E1基因第6内含子上,可被限制性内切酶DraⅠ识别的位点被命名为CYP2E1*6,其基因型表现为:野生型TT、杂合突变型TA和纯合突变型AA。位于CYP2E1基因5′端调控序列上,96个碱基对插入/缺失的多态性被称为CYP2E1*1D/*1C,对酶活性也具有调节作用[8]。
在INH代谢过程中,CYP2E1可将乙酰烟肼转化为乙酰偶氮、烯酮和乙酰阳离子等肝毒性物质[3],CYP2E1酶活性升高时,肝毒性物质生成增多,将增加ATDH风险[8]。研究者对CYP2E1基因c1/c1基因型与ATDH易感性的相关性进行了Meta分析(纳入13项病例对照研究),结果提示:在东亚人群中,CYP2E1基因c1/c1基因型与ATDH易感性具有相关性,但在印度人群、高加索人群及其他人群中,两者的相关性无统计学意义[11]。尽管Meta分析的结果提示,在中国人群中,CYP2E1基因c1/c1基因型与ATDH患病风险具有相关性[12];但3项独立研究的结果均提示,CYP2E1基因c1/c1基因型与中国人群ATDH患病风险并无相关性[13~15]。虽然CYP2E1基因DraⅠ位点多态性与ATDH患病风险的相关性研究较少,但却存在争议。在印度人群中,2项独立研究分别发现CYP2E1基因DraⅠ位点CC基因型或CD基因型与ATDH患病风险具有相关性[16, 17],但在中国人群中,Tang等[13]并未发现CYP2E1基因DraⅠ位点多态性与ATDH患病风险的相关性。
3.1.1.2CYP3A4是肝内最丰富的CYP亚家族,参与临床上50%以上药物的代谢[18]。CYP3A4与卡马西平、INH等多种药物引起的细胞毒性有关[18],而CYP3A4抑制剂可显著减弱INH的细胞毒性作用[19]。而且近年来的研究提示,CYP3A4对INH的代谢、生物活化和肝毒性均有影响[19]。CYP3A4对INH致肝毒性的影响可能与孕烷X受体(PXR)有关,RMP通过激活PXR诱导CYP3A4的表达,激活的CYP3A4可促进INH产生肝毒性代谢产物[3]。动物实验研究并未发现CYP3A4对小鼠体内INH药代动力学的影响[19]。在巴西人群中,研究者也未发现CYP3A4基因-392位点A>G变异与ATDH风险的显著相关性,可能与该研究人群大部分为CYP3A4基因-392位点AA基因型携带者有关[20]。
3.1.1.3CYP2B6是CYP家族中重要的药物代谢酶,参与多种内源性和外源性物质的合成和代谢[21]。RMP可通过诱导CYP2B6表达,使INH肝毒性代谢产物增多,增加ATDH风险[22]。CYP2B6基因多态性与ATDH患病风险的相关性研究,多集中在TB- HIV共感染人群。高活性抗逆转录病毒疗法(HAART)药物依法韦仑,主要通过CYP2B6代谢[23]。研究人员发现,在TB-HIV共感染人群中,CYP2B6基因516位点TT基因型与ATDH患病风险的相关性存在争议,在坦桑尼亚人群[23]、埃塞俄比亚人群[24]中,CYP2B6基因516位点TT基因型是ATDH发生的危险因素;但在巴西人群中,CYP2B6基因516位点TT基因型是ATDH进展的保护性因素[22]。
3.1.1.4CES1是丝氨酸酯酶超家族成员之一,此家族成员主要催化含酯、硫酯和酰胺键的内源性和外源性物质的水解或酯交换反应[2]。人类基因组中最具特征性的CES为CES1、CES2和CES3,均在肝内表达[25]。CES1和CES2在肝内高表达,与INH代谢密切相关[25];CES3在脑血管内皮细胞中高表达,与血脑屏障功能有关[25];CES4是位于CES1上游28 kb处的转录假基因,与CES1高度相似[25]。如图1所示,酰胺酶在INH代谢过程中,通过酰胺键裂解作用水解INH[26],动物实验研究也证实酰胺酶活性对肝毒性有影响[27]。因此,肝内具有酰胺酶裂解作用的CES,可能主要通过干扰INH代谢影响ATDH风险[25]。
在英国人群中,Yamada等[25]纳入170例潜伏结核分枝杆菌感染患者,对CES1、CES2、CES4所有外显子和启动子序列进行检测,结果提示,所有检测的单核苷酸多态性(SNPs)与ATDH患病风险均无显著相关性,只有CES1基因-2位点C/G变异改变了CES1基因的翻译起始序列。在中国人群中,Wu等[28]发现CES1基因rs8192950-AC基因型和rs1968785-GG基因型是ATDH的保护性因素。
3.1.2Ⅱ相代谢酶
3.1.2.1NAT2如图1所示,NAT2是INH代谢过程中的主要代谢酶,其基因多态性影响NAT2酶活性[5]。一般来说,2个慢乙酰化等位基因(NAT2*5、NAT2*6、NAT2*7、NAT2*14)组成的基因型定义为慢乙酰化表型,1个慢和1个快乙酰化等位基因(NAT2*4、NAT2*11、NAT2*12、NAT2*13)组成的基因型定义为中间乙酰化表型,2个快乙酰化等位基因组成的基因型定义为快乙酰化表型[5, 29]。在NAT2酶作用下,在快乙酰化表型者体内,INH通过主路途径迅速转化为二乙酰肼并排出体外;而在慢乙酰化表型者体内,由于INH主路途径代谢减慢而旁路代谢途径相对增强,INH直接水解成肼的量增加[6]。同时,慢乙酰化表型者不能将乙酰烟肼迅速转化为二乙酰肼,而增加了CYP2E1途径的代谢,产生的肝毒性物质增多[6]。NAT2代谢表型多态性分布具有地域特点,亚洲国家以快代谢表型为主,而欧美及非洲国家则主要以中间/慢代谢表型为主[30]。本课题组的研究结果提示,在中国汉族儿童中,NAT2代谢表型以快代谢表型的频率最高(61.3%),其次是中间/慢代谢表型(34.1%)[31]。
尽管既往研究认为快乙酰化者更易发生ATDH,但近年来多项研究指出,与快乙酰化表型者相比,慢乙酰化表型者更易发生ATDH,而且症状更严重[3, 5]。一项纳入24项病例对照研究的Meta分析结果提示,在不同人群(东亚人群、印度人群、中东人群及其他人群)中,NAT2慢乙酰化表型者更易发生ATDH,但在高加索人群中,研究者并未发现NAT2基因多态性与ATDH易感性的相关性[11]。中国台湾[15]及北京[14]的研究结果均提示,在中国人群中,NAT2慢乙酰化表型者更易发生ATDH;但在中国社区人群中,Lv等[32]并未发现NAT2基因型与ATDH易感性的相关性。
3.1.2.2GST是通过催化多种物质的毒性中间代谢产物与GSH结合,并排出体外而起到解毒作用,与多种致癌物、毒性化学物和药物的解毒有关[2, 8]。GST至少由5个独立位点编码,即α、μ、π、θ、γ,在肝毒性相关研究中,GSTM1(μ型)和GSTT1(θ型)获得较多关注[8]。在人群中,携带1个或2个GSTM1和GSTT1功能性等位基因的个体具有酶活性,而纯合子缺失基因型个体酶活性丧失[8]。报道显示,在不同人群健康个体中,GSTM1和GSTT1纯合子缺失基因型频率差异较大,在太平洋岛民、马来西亚人群、中国人群、韩国人群、日本人群中,GSTM1和GSTT1纯合子缺失基因型频率相对较高[8]。本课题组的研究结果提示,在中国汉族儿童中,GSTM1和GSTT1纯合子缺失基因型的频率最高,分别为59.3%、58.4%,单拷贝缺失基因型的频率次之,分别为34.0%、35.1%,未缺失基因型的频率最低,分别为6.7%、6.5%[33]。
GSTM1和GSTT1纯合子缺失基因型携带者的GST酶活性丧失,对INH毒性中间代谢产物的解毒能力降低,可能易发生ATDH[3]。一项纳入11项病例对照研究的Meta分析结果提示,在东亚人群中,GSTM1缺失基因型与ATDH风险具有相关性,但在印度人群和高加索人群中,两者的相关性并不显著[11]。有Meta分析[34]提示,在中国人群中,GSTM1缺失基因型与ATDH患病风险具有显著相关性,但Tang等[13]及本课题组[35]在中国人群中并未发现两者的相关性。虽然体内、体外研究均提示,GSTT1纯合子缺失基因型与GST酶活性丧失有关[34],上述2项独立Meta分析[11, 34]结果均提示,在所有人群(高加索人群、印度人群、东亚人群)中,GSTT1缺失基因型与ATDH风险并无相关性。
3.1.2.3UGTUGT家族是位于内质网的膜结合酶,催化葡萄糖醛酸基与亲核性物质结合,与多种内外源性物质的解毒和消除有关[5]。UGT家族可分为2个亚家族,分别由UGT1A和UGT2编码[2]。UGT1A由9个活性基因组成,如UGT1A1、UGT1A3、UGT1A6;UGT2进一步被分为UGT2A和UGT2B[2]。
在不同人群中,UGT基因多态性与ATDH患病风险的相关性研究较少。在韩国人群中,Kim等[36]并未发现UGT1A1基因多态性与ATDH患病风险的相关性,但在中国台湾人群中,Change等[37]发现同时携带UGT1A1*27和UGT1A1*28等位基因的个体更易发生ATDH 。UGT1A3基因多态性与韩国人群ATDH患病风险并无相关性[36]。在中国人群中,Hao等[38]发现UGT1A6基因-19位点T/G变异、-308位点C/A变异、-541位点C/A变异与ATDH患病风险有关。在埃塞俄比亚TB- HIV共感染患者中并未发现UGT2B7基因-372位点G>A变异与ATDH患病风险的相关性[24];但在中国人群中发现UGT2B7基因-268位点AG基因型和802位点TT基因型携带者,发生ATDH风险明显高于野生型(UGT2B7基因-268位点AA基因型和802位点CC基因型)携带者[39]。
3.1.2.4MnSOD摄入体内的药物在代谢过程中产生过量ROS,可引起脂质过氧化和细胞死亡[3]。MnSOD可将线粒体内产生的ROS催化还原为过氧化氢,是抗氧化应激的关键酶[2]。MnSOD基因47位点的多态性与许多癌症和酒精性肝疾病风险增加有关,研究人员认为MnSOD基因47位点发生T/C置换后,导致前导氨基酸序列上缬氨酸转变为丙氨酸,改变了线粒体靶向序列的螺旋结构,使得进入线粒体基质的MnSOD增多,催化生成的过氧化氢也增多,造成其后的解毒负荷过大,易发生肝损伤[5, 7]。中国台湾人群MnSOD基因47位点TC或CC基因型携带者发生ATDH风险增加[40],中国北京研究也显示,MnSOD基因47位点CC基因型与ATDH进展具有显著相关性[41]。MnSOD基因47位点变异与ATDH风险仅在中国人群中观察到相关性,因此,未来需要进行多种族、大样本的相关研究。
3.2药物转运体(Ⅲ相代谢酶) 与抗结核药物代谢相关的药物转运体主要为肝脏转运体,分为基质侧膜转运体和胆管侧膜转运体。前者主要调控肝细胞从肝血窦摄取物质,主要是SLC转运体;后者主要调控肝细胞将物质排入胆汁内,主要是ABC转运体[2, 42]。
3.2.1SLC转运体主要调控肝细胞摄取肝血窦中外源性化合物和结合型胆汁酸,分为钠依赖性途径和非钠依赖性途径;前者主要是牛磺胆酸钠共转运多肽(NTCP,由SLC10A1编码),后者主要是有机阴离子转运蛋白(OAT)和有机阴离子转运多肽(OATP,由SLCO1B1编码)家族的成员[2, 42]。
OATP与肝细胞从肝血窦摄取药物(如RMP)有关,SLCO1B1基因多态性可影响药物摄取的药代动力学[43]。在亚洲人群中,rs2306283(388A>G)、rs4149056(521T>C)突变频率较高,而且两者存在连锁不平衡,组成的4种单倍体分别是SLCO1B1*1A(388A-521T)、*1B(388G-521T)、*5(388A-521C)和*15(388G-521C)[43]。在韩国人群[44]、印度人群[45]及埃塞俄比亚人群[24]中,研究者并未发现SLCO1B1基因多态性与ATDH患病风险的相关性;但在中国人群发现SLCO1B1基因521位点T>C变异[46]、SLCO1B1 *15单倍体[43]与ATDH患病风险增加有关。目前,进行SLC10A1基因多态性与ATDH患病风险的相关研究较少,而且在中国人群未发现SLCO10A1基因多态性与ATDH患病风险的相关性[43]。
3.2.2ABC转运体利用水解ATP的能量,将肝细胞内的药物分子和代谢产物泵出胞内。ABC转运体可分为7个亚族,在药物分布中具有重要作用的主要是多药耐药蛋白1(MDR1,又称P-糖蛋白,由ABCB1编码)、多药耐药相关蛋白2(MRP2,由ABCC2编码)和乳腺癌耐药蛋白(BCRP,由ABCG2编码)[2]。
在韩国人群[44]、中国人群[46]并未发现ABCB1基因多态性与ATDH患病风险的相关性;但在埃塞俄比亚TB-HIV共感染人群中,ABCB1基因3435位点TT基因型与ATDH患病风险增加有关[24]。在韩国人群[44]、埃塞俄比亚人群[24]未发现ABCC2基因多态性与ATDH患病风险的相关性。
3.3抗氧化反应药物代谢产生的ROS或活性代谢产物可引起抗氧化反应,参与抗氧化反应的代谢酶主要有GST和MnSOD以及Nrf-ARE信号通路。
在肝细胞内,抗结核药物代谢产生毒性中间代谢产物,其在CYP2E1等催化下产生过量ROS和活性氮(RNS)[9]。肝内ROS/RNS存在时,核因子活化B细胞κ轻链增强子(NF-κB)上调诱导型一氧化氮合酶(iNOS,由NOS2A编码)表达,产生过量肝毒性分子一氧化氮[9]。机体处于生理状态时,体内多种抗氧化酶可快速消除ROS/RNS,如GSTs、NAD(P)醌脱氢酶1(NQO1,由NQO1编码)、血红素加氧酶(HO1,由HMOX1编码)等[9]。多种转录因子可调节抗氧化酶基因启动子区域上的抗氧化反应原件(ARE),如Nrf2(由NFE2L2编码)、BTB-CNC异体同源体1(Bach1,由BACH1编码)、小Maf碱性亮氨酸拉链蛋白(small Maf basic leucine zipper proteins,主要是MafF、MafG、MafK,分别由MAFF、MAFG、MAFK编码)[9]。如图2所示, 在细胞核内,
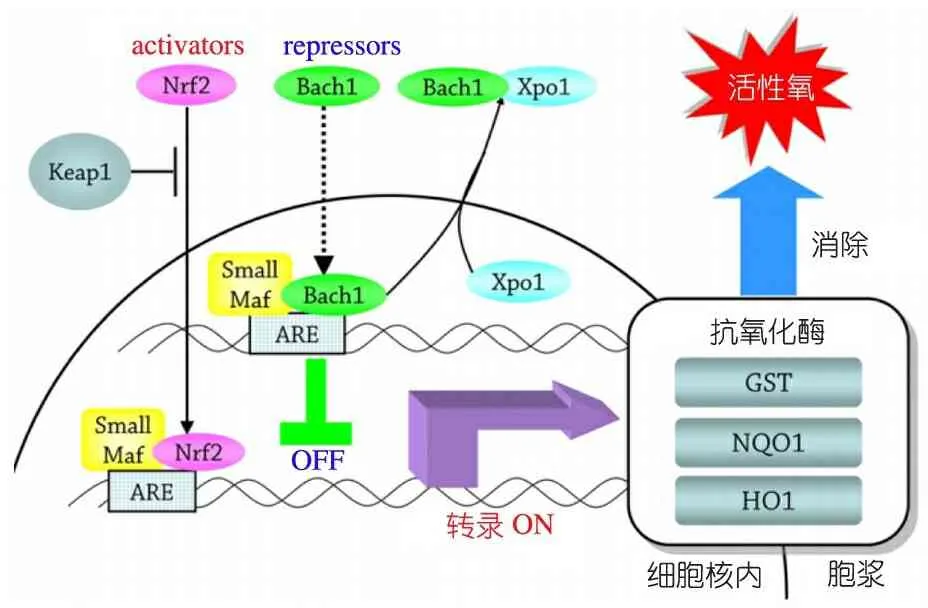
图2INrf-ARE信号通路[9]
注Nrf2:核因子2相关因子2;ARE:抗氧化反应元件;Keap1:Kelch样环氧氯丙烷相关蛋白1;Small Maf:小Maf碱性亮氨酸拉链蛋白;Bach1:BTB-CNC异体同源体1;Xpo1:输出蛋白1;GST:谷胱甘肽S转移酶;NQO1:NAD(P)H醌脱氢酶1;HO1:血红素加氧酶1
Nrf2和小Maf蛋白形成的异源二聚体与ARE结合,上调抗氧化酶表达[3];相反,Bach1和小Maf蛋白形成的异源二聚体与ARE结合,下调抗氧化酶表达[3]。在非氧化应激状态时,胞浆中的Kelch样环氧氯丙烷相关蛋白1(Keap1,由KEAP1编码)与Nrf2结合,抑制Nrf2进入核内,从而抑制抗氧化酶表达[9];相反,输出蛋白1(Xpo1,由XPO1编码)与Bach1结合,将其从核内运到胞浆,从而激活抗氧化酶表达[9]。因此,体内Nrf-ARE信号通路中,抗氧化激活途径(包括Nrf2/小Mafs/Xpo1)和抑制途径(包括Bach1/小Mafs/Keap1)的失调可能导致ATDH的发生[3]。
Nanashima等[9]对日本人群Nrf-ARE信号通路中10个基因的多态性与ATDH患病风险的相关性进行分析,结果提示,在所检测的34个标签SNPs中,只有3个SNPs是发生ATDH的独立危险因素,分别是NOS2A基因rs11080344-CC基因型、BACH1基因rs2070401-CC基因型以及MAFK基因rs4720833-GA或AA基因型。
3.4免疫反应从ATDH发病机制可知,免疫反应在ATDH发生过程中发挥了重要作用。体内炎症-抗炎反应的失衡方向,决定肝细胞是发生损伤反应还是修复反应[3],而TNF-α、IL-4、IL-6和IL-10对炎症-抗炎平衡影响较大。在人体内,TNF-α启动子转录起始位点上游308核苷酸G/A转变与TNF-α水平升高有关[2];Kim等[47]发现,TNF-α基因-308位点G/A变异与ATDH的发生具有相关性。IL-4基因多态性与药物性肝损伤(DIH)易感性具有相关性[2],但在TB人群中,IL-4基因多态性与ATDH易感性无相关性[48]。IL-6与急性期反应基因的调节有关[4],Wang等[49]未发现IL-6基因多态性与ATDH易感性的相关性。动物实验显示IL-10与DIH有关[2],但人群研究并未发现IL-10基因多态性与ATDH易感性的相关性[48]。
MHC主要负责将抗原提呈给T细胞以激活保护性免疫反应[2, 3],人类MHC通常被称为人类白细胞抗原(HLA)。HLA分子主要有2种,即Ⅰ类(HLA-A、HLA-B及HLA-C)和Ⅱ类(HLA-DP、HLA-DQ及HLA-DR)[4];其中HLA-DQ基因具有高度多态性,其基因多态性可改变HLA-DQ分子的功能[50]。Sharma等[51]发现印度人群HLA-DQA1*0102等位基因缺失和HLA-DQB1*0201等位基因存在是ATDH发生的独立危险因素;Chen等[50]发现,中国人群HLA-DQB1*05/*05基因型与ATDH患病风险具有相关性。
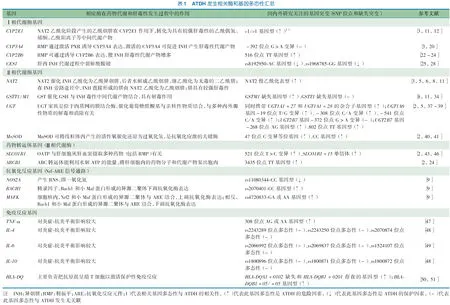
与ATDH发生相关酶和基因多态性汇总见表1。
4 其他因素对ATDH的影响
年龄、性别、营养状态、酒精摄入和肝脏基础疾病等非遗传因素,也有可能对ATDH患病风险产生影响。
4.1年龄是影响ATDH发生的重要因素[2, 3]。儿童是ATDH的高危人群[52]。接受INH和RMP治疗后,儿童比成人更易发生ATDH(6.9%vs2.7%)[3],可能与儿童的代谢酶与器官(如肝脏、肾脏)发育不成熟,药物代谢过程不完善、清除率降低,药物和毒性代谢产物易在体内累积有关。老年患者也是ATDH的高危人群[2, 3]。近期的研究提示,与<60岁TB患者相比,≥60岁TB患者的ATDH风险增加了2.6~3.5倍[3];而且,≥50岁TB患者ATDH的严重程度及病死率均高于<50岁TB患者[3]。可能与老年患者DME活性降低、肝功能恶化有关[2]。
4.2性别多项研究显示,女性患ATDH风险高于男性[3, 6, 7, 52],而且女性比男性更易发生严重肝疾病[52],可能与女性CYP3A酶活性高于男性有关[3, 6]。CYP3A4对INH代谢的影响可能受PXR调控[3]。Wang等[53]发现,中国人群PXR基因rs2461823位点和rs7643645位点的多态性与ATDH易感性具有相关性,但这种相关性只特异地存在于女性人群。
4.3营养状态TB患者容易发生营养不良,而白蛋白低(<35 g·L-1)的患者[52]和4周内体重减轻>2 kg患者[3, 5, 52],ATDH患病风险明显增加。可能与CYP450酶系统受营养摄入、空腹、营养不良状态的影响[3],导致药物清除率降低、血药浓度升高[6]有关。
4.4酒精摄入酒精通过诱导CYP2E1等肝酶,增加患者ATDH患病风险[2, 3, 5]。饮酒过量(每日酒精摄入量超过48 g)者,ATDH患病风险是非饮酒过量者的3倍[54];饮酒甚至使进行RMP预防性治疗的TB患者,ATDH患病风险增加[3]。
4.5肝脏基础疾病一般来说,肝功能检测基线异常的患者肝毒性发生率更高[2]。TB-病毒联合感染的轻度炎症反应使机体处于高危状态,使用抗结核药物后,TB-病毒联合感染者易从起始高危状态进入全面肝毒性反应[3]。与未感染者相比,感染乙型肝炎病毒(HBV)或丙型肝炎病毒(HCV)的TB患者发生ATDH风险更高,而且肝毒性严重程度与抗结核治疗前起始病毒载量直接相关[3]。与未感染HIV者相比,HIV感染者更易进展为活性TB[52],TB-HIV共感染者ATDH发生风险也显著增加[3];而且,HAART治疗前后,TB-HIV共感染者ATDH发生风险均显著增加[3]。
4.6吸烟烟草烟雾中的不同成分可通过不同途径对肝脏产生毒性作用,如多环芳香烃、尼古丁等通过诱导CYP1A1、CYP1A2、CYP2E1等而干扰药物清除[55],丙烯醇通过线粒体毒性作用,使线粒体功能紊乱[56]。然而也有学者认为,烟草烟雾中的杂环胺需要CYP1A2和NAT2激活,长期吸烟可增加慢乙酰化代谢表型者体内的NAT2酶活性,其体内INH代谢反而加快[57]。Camila等[20]发现,长期吸烟是巴西人群ATDH进展的保护性因素。
5 其他
给药方案、病变类型以及药物间相互作用,对ATDH患病风险也有一定影响。每日用药的抗结核治疗方案,ATDH患病风险高于每周3次用药的抗结核治疗方案[6];血行播散型TB的ATDH发生率较高[58];INH与RMP、PZA合用时 , 比INH单用时ATDH的发生率高[2, 3], 抗结核药物与HAART同时进行时,ATDH风险高于2种治疗方法单独使用时[3, 52]。
6 小结
ATDH现在是而且将来仍是全球严重的ADR。DME、药物转运体、抗氧化反应及免疫反应多种基因的多态性与ATDH易感性有关,而且年龄、性别、酒精摄入、肝脏基础疾病和吸烟等多种非遗传因素,对ATDH患病风险也有一定影响。因此,了解与ATDH易感性相关的基因多态性和非遗传危险因素,有助于识别ATDH高危患者及预测ATDH的发生。对于已识别的高危基因型与高危因素的患者,需在抗结核药物治疗的前几个月定期进行肝生化检验。除此之外,仍需开展ATDH易感基因多态性方面的大样本、多种族、多中心的风险评估研究,调整不同基因型个体给药剂量以预防ATDH的发生。
[1]Frieden T, Sterling T, Munsiff S, et al. Tuberculosis. Lancet, 2003,362:887-899
[2]Chen R, Wang J, Zhang Y, et al. Key factors of susceptibility to anti-tuberculosis drug-induced hepatotoxicity. Arch Toxicol, 2015,89(6):883-897
[3]Ramappa V, Aithal GP. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J Clin Exp Hepatol, 2013,3(1):37-49
[4]Daly AK, Day CP. Genetic association studies in drug-induced liver injury. Drug Metab Rev, 2012,44(1):116-126
[5]Huang YS. Recent progress in genetic variation and risk of antituberculosis drug-induced liver injury. J Chin Med Assoc, 2014,77(4):169-173
[6]Tostmann A, Boeree MJ, Aarnoutse RE, et al. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J Gastroenterol Hepatol, 2008,23(2):192-202
[7]Corsini A, Bortolini M. Drug-induced liver injury: the role of drug metabolism and transport. J Clin Pharmacol, 2013,53(5):463-474
[8]Roy PD, Majumder M, Roy B. Pharmacogenomics of anti-TB drugs-related hepatotoxicity. Pharmacogenomics, 2008,9(3):311-321
[9]Nanashima K, Mawatari T, Tahara N, et al. Genetic variants in antioxidant pathway: risk factors for hepatotoxicity in tuberculosis patients. Tuberculosis (Edinb), 2012,92(3):253-259
[10]Adam J, Pichler WJ, Yerly D. Delayed drug hypersensitivity: models of T-cell stimulation. Br J Clin Pharmacol, 2011,71(5):701-707
[11]Cai Y, Yi J, Zhou C, et al. Pharmacogenetic study of drug-metabolising enzyme polymorphisms on the risk of anti-tuberculosis drug-induced liver injury: a meta-analysis. PLoS One, 2012,7(10):e47769
[12]Deng R, Yang T, Wang Y, et al. CYP2E1 RsaI/PstI polymorphism and risk of anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis, 2012,16(12):1574-1581
[13]Tang SW, Lv XZ, Zhang Y, et al. CYP2E1, GSTM1 and GSTT1 genetic polymorphisms and susceptibility to antituberculosis drug-induced hepatotoxicity: a nested case-control study. J Clin Pharm Ther, 2012,37(5):588-593
[14]An HR, Wu XQ, Wang ZY, et al. NAT2 and CYP2E1 polymorphisms associated with antituberculosis drug-induced hepatotoxicity in Chinese patients. Clin Exp Pharmacol Physiol, 2012,39(6):535-543
[15]Lee SW, Chung LS, Huang HH, et al. NAT2 and CYP2E1 polymorphisms and susceptibility to first-line anti-tuberculosis drug-induced hepatitis. Int J Tuberc Lung Dis, 2010,14(5):622-626
[16]Roy B, Ghosh SK, Sutradhar D, et al. Predisposition of antituberculosis drug induced hepatotoxicity by cytochrome P450 2E1 genotype and haplotype in pediatric patients. J Gastroenterol Hepatol, 2006,21(4):784-786
[17]Bose PD, Sarma MP, Medhi S, et al. Role of polymorphic N-acetyl transferase2 and cytochrome P4502E1 gene in antituberculosis treatment-induced hepatitis. J Gastroenterol Hepatol, 2011,26(2):312-318
[18]Yoshikawa Y, Hosomi H, Fukami T, et al. Establishment of knockdown of superoxide dismutase 2 and expression of CYP3A4 cell system to evaluate drug-induced cytotoxicity. Toxicol In Vitro, 2009,23(6):1179-1187
[19]Liu K, Li F, Lu J, et al. Role of CYP3A in isoniazid metabolism in vivo. Drug Metab Pharmacokinet, 2014,29(2):219-222
[20]Zaverucha-do-Valle C, Monteiro SP, El-Jaick KB, et al. The role of cigarette smoking and liver enzymes polymorphisms in anti-tuberculosis drug-induced hepatotoxicity in Brazilian patients. Tuberculosis (Edinb), 2014,94(3):299-305
[21]Miksys S, Lerman C, Shields PG, et al. Smoking, alcoholism and genetic polymorphisms alter CYP2B6 levels in human brain. Neuropharmacology, 2003,45(1):122-132
[22]Fernandes DC, Santos NP, Moraes MR, et al. Association of the CYP2B6 gene with anti-tuberculosis drug-induced hepatotoxicity in a Brazilian Amazon population. Int J Infect Dis, 2015,33:28-31
[23]Mugusi S, Ngaimisi E, Janabi M, et al. Liver enzyme abnormalities and associated risk factors in HIV patients on efavirenz-based HAART with or without tuberculosis co-infection in Tanzania. PLoS One, 2012,7(7):e40180
[24]Yimer G, Ueda N, Habtewold A, et al. Pharmacogenetic & pharmacokinetic biomarker for efavirenz based ARV and rifampicin based anti-TB drug induced liver injury in TB-HIV infected patients. PLoS One, 2011,6(12):e27810
[25]Yamada S, Richardson K, Tang M, et al. Genetic variation in carboxylesterase genes and susceptibility to isoniazid-induced hepatotoxicity. Pharmacogenomics J, 2010,10(6):524-536
[26]Kita T, Tanigawara Y, Chikazawa S, et al. N-Acetyltransferase2 genotype correlated with isoniazid acetylation in Japanese tuberculous patients. Biol Pharm Bull, 2001,24(5):544-549
[27]Sarich TC, Adams SP, Petricca G, et al. Inhibition of isoniazid-induced hepatotoxicity in rabbits by pretreatment with an amidase inhibitor. J Pharmacol Exp Ther, 1999,289(2):695-702
[28]吴雪琼,朱冬林,张俊仙,等. 羧酸酯酶基因1多态性鉴定及其与抗结核药物肝毒性相关性研究. 中华内科杂志, 2012,51(7):524-530
[29]Hein DW, Doll MA. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics, 2012,13(1):31-41
[30]Sabbagh A, Langaney A, Darlu P, et al. Worldwide distribution of NAT2 diversity: implications for NAT2 evolutionary history. BMC Genet, 2008,9:21
[31]刘芳, 苗青, 焦伟伟, 等. NAT2和CYP2E1基因多态性及其代谢表型在中国汉族儿童中的分布. 中国当代儿科杂志, 2012(05):353-358
[32]Lv X, Tang S, Xia Y, et al. NAT2 genetic polymorphisms and anti-tuberculosis drug-induced hepatotoxicity in Chinese community population. Ann Hepatol, 2012,11(5):700-707
[33]刘芳, 苗青, 肖婧, 等. 汉族儿童Ⅱ相药物代谢酶基因GSTM1和GSTT1的多态性分布. 中国循证儿科杂志, 2011(03):205-210
[34]Tang N, Deng R, Wang Y, et al. GSTM1 and GSTT1 null polymorphisms and susceptibility to anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis, 2013,17(1):17-25
[35]Liu F, Jiao AX, Wu XR, et al. Impact of glutathione S-transferase M1 and T1 on anti-tuberculosis drug-induced hepatotoxicity in Chinese pediatric patients. PLoS One, 2014,9(12):e115410
[36]Kim SH, Kim SH, Bahn JW, et al. Genetic polymorphisms of drug-metabolizing enzymes and anti-TB drug-induced hepatitis. Pharmacogenomics, 2009,10(11):1767-1779
[37]Chang JC, Liu EH, Lee CN, et al. UGT1A1 polymorphisms associated with risk of induced liver disorders by anti-tuberculosis medications. Int J Tuberc Lung Dis, 2012,16(3):376-378
[38]郝金奇,陈怡,李世明,等. 尿苷二磷酸葡萄糖醛酸转移酶1A6基因多态性与抗结核药致肝损害的相关性. 中华肝脏病杂志, 2011,19(3):201-204
[39]史哲, 贺蕾, 高丽, 等. UGT2B7基因多态性与抗结核药物性肝损伤的相关性分析. 中国抗生素杂志, 2014(11):856-860
[40]Huang YS, Su WJ, Huang YH, et al. Genetic polymorphisms of manganese superoxide dismutase, NAD(P)H:quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J Hepatol, 2007,47(1):128-134
[41]An HR, Wu XQ, Wang ZY. The relationship between the polymorphism of MnSOD gene and antituberculosis drug-induced liver injury. Chinese Journal of Antibiotics, 2012,37(11):1-4
[42]Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol, 2013,58(1):155-168
[43]Chen R, Wang J, Tang S, et al. Association of polymorphisms in drug transporter genes (SLCO1B1 and SLC10A1) and anti-tuberculosis drug-induced hepatotoxicity in a Chinese cohort. Tuberculosis (Edinb), 2015,95(1):68-74
[44]Kim SH, Kim SH, Lee JH, et al. Polymorphisms in drug transporter genes (ABCB1, SLCO1B1 and ABCC2) and hepatitis induced by antituberculosis drugs. Tuberculosis (Edinb), 2012,92(1):100-104
[45]Singh M, Gupta VH, Amarapurkar DN, et al. Association of genetic variants with anti-tuberculosis drug induced hepatotoxicity: a high resolution melting analysis. Infect Genet Evol, 2014,23:42-48
[46]张金玲, 朱学彬, 李世明, 等. SLCO1B1/ABCB1基因多态性与抗结核药物性肝损伤的相关性分析. 中华疾病控制杂志, 2013(06):469-472
[47]Kim SH, Kim SH, Yoon HJ, et al. TNF-alpha genetic polymorphism -308G/A and antituberculosis drug-induced hepatitis. Liver Int, 2012,32(5):809-814
[48]Wang J, Chen R, Tang S, et al. Interleukin-4 and interleukin-10 polymorphisms and antituberculosis drug-induced hepatotoxicity in Chinese population. J Clin Pharm Ther, 2015,40(2):186-191
[49]Wang J, Chen R, Tang S, et al. Analysis of IL-6, STAT3 and HSPA1L gene polymorphisms in anti-tuberculosis drug-induced hepatitis in a nested case-control study. PLoS One, 2015,10(3):e118862
[50]Chen R, Zhang Y, Tang S, et al. The association between HLA-DQB1 polymorphism and antituberculosis drug-induced liver injury: a Case-Control Study. J Clin Pharm Ther, 2015,40(1):110-115
[51]Sharma SK, Balamurugan A, Saha PK, et al. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am J Respir Crit Care Med, 2002,166(7):916-919
[52]Devarbhavi H. Antituberculous drug-induced liver injury: current perspective. Trop Gastroenterol, 2011,32(3):167-174
[53]Wang JY, Tsai CH, Lee YL, et al. Gender-Dimorphic Impact of PXR Genotype and Haplotype on Hepatotoxicity During Antituberculosis Treatment. Medicine (Baltimore), 2015,94(24):e982
[54]Sharma SK, Balamurugan A, Saha PK, et al. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am J Respir Crit Care Med, 2002,166(7):916-919
[55]Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm, 2007,64(18):1917-1921
[56]Sun L, Luo C, Long J, et al. Acrolein is a mitochondrial toxin: effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion, 2006,6(3):136-142
[57]Voutsinas J, Wilkens LR, Franke A, et al. Heterocyclic amine intake, smoking, cytochrome P450 1A2 and N-acetylation phenotypes, and risk of colorectal adenoma in a multiethnic population. Gut, 2013,62(3):416-422[58]Donald PR. Antituberculosis drug-induced hepatotoxicity in children. Pediatr Rep, 2011,3(2):e16
(本文编辑:张崇凡)
10.3969/j.issn.1673-5501.2016.04.013
国家自然科学基金应急管理项目:81441067;国家自然科学基金面上项目:81571950;北京市科委行业定额经费资助:2014-bjsekyjs-1;北京市科技新星计划交叉学科合作课题:xxjc2001611;2015年北京市留学人员科技活动择优资助项目
首都医科大学附属北京儿童医院、北京市儿科研究所,儿科学国家重点学科,教育部儿科重大疾病研究重点实验室,国家呼吸系统疾病临床医学研究中心,儿童呼吸道感染性疾病研究北京市重点实验室北京,100045
申阿东,E-mail:shenad16@hotmail.com
2016-05-15
2016-08-20)
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
昆明医科大学学报(2021年4期)2021-07-23 01:22:00
上海工运(2020年8期)2020-12-14 03:11:56
科学(2020年3期)2020-11-26 08:18:20
中国民族医药杂志(2016年4期)2016-05-09 07:41:11
工业设计(2016年4期)2016-05-04 04:00:14
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
中国医药生物技术(2015年4期)2015-12-26 08:26:36
结核与肺部疾病杂志(2015年4期)2015-07-18 11:08:22
