
产褥期阴道微生态研究
2016-09-13 00:30张群英王黎陈祖云陈欢方序
浙江临床医学 2016年3期
张群英 王黎 陈祖云 陈欢⋆ 方序
产褥期阴道微生态研究
张群英 王黎 陈祖云 陈欢⋆ 方序
目的 应用细菌基因测序技术了解阴道内微生物种群的分布情况。方法 采集10例产褥期妇女阴道分泌物(pH值、清洁度、脓球、滴虫、霉菌等)测定雌激素,提取微生物总基因组,扩增16S rRNA 测序。另选择正常育龄妇女为对照,比较两组结果了解产褥期阴道微生态情况,菌种组成情况和生理情况。结果 正常妇女阴道中的乳杆菌属(Lactobacillus)的含量是产褥期妇女的>100倍,早期产妇,乳酸杆菌显著下降,这导致其抵抗力下降。不动杆菌属在产妇早期的的阴道中含量上升,表明其抵抗力下降。结论 产褥期孕妇的微生态呈现一个动态的过程,早期产褥妇女与正常妇女有显著差异,而在产褥后期的微生态基本与正常妇女一致。16S rRNA测序可以较好的揭示阴道微生态的变化,其有希望成为一个新的、系统的评估手段。不仅可以分析产妇阴道微生态的恢复情况,还能用于检测和评估其综合情况。
阴道微生态 16S rRNA 高通量测序 乳酸杆菌 不动杆菌
正常阴道内有多种细菌存在,这种微生态的形成对阻止泌尿生殖系统疾病的发生起着至关重要的作用,如乳酸杆菌维持阴道正常的酸性环境,抑制其它病原体生长。研究发现,产褥期妇女阴道微生态失调占75.31%,阴道炎占9.1%[1]。阴道微生态的评价方法中,宏基因技术相比传统的培养有明显的优越性[2]。为更好的了解我国妇女在产褥期阴道微生态的变化,作者采集产褥期妇女和正常育龄妇女的阴道分泌物,应用细菌基因测序技术以期了解阴道内微生物种群的分布情况。
1 临床资料
1.1 一般资料 收集2013年9月至2014年11月本院10例产褥期妇女(产后6~8周)阴道分泌物(标记为T)为观察组,年龄25~38岁,平均年龄33.5岁。以6周为界,<42d为产褥期早期(6例),42~56d为产褥期晚期(4例)。另收集正常育龄妇女的阴道分泌物(10例,标记为N)为对照组,年龄21~47岁,平均年龄35岁。纳入标准:所有入选妇女近<3d均无性生活;无阴道分泌物异常、接触性出血;无宫颈手术史、无妊娠;无盆腔放射治疗史;年龄差异无统计学意义。
1.2 方法 (1)标本采集:所有妇女均用无菌刮板及长棉签从阴道上1/3处获取阴道分泌物。(2)无菌刮板检测pH值、清洁度、脓球、滴虫、霉菌等检查和雌激素测定。(3)DNA提取:长棉签采集的微生物悬浮于0.5ml灭菌PBS溶液中,加入50μl溶菌酶(10mg/ml),6μl变溶菌素(25000 U/ ml;Sigma-Aldrich),3μl溶葡球菌酶(4000 U/ ml;Sigma-Aldrich),和41μl TE50缓冲液(10 mM Tris·HCL和50 mM EDTA,pH 8.0),37℃消化1h。然后利用细菌基因组提取试剂盒(Cat No. D-6000A,Gentra,USA)提取DNA。(4)高通量16S rDNA测序: 对细菌的V4区,利用Miseq进行测序,扩增引物为:F:5'-AYTGGGYDTAAAGNG-3',R:5'-TACNVGGGTATCTAATCC-3',采用Illumina MiSeq测序平台进行。(5)测序结果的分析:测序所得结果根据不同的Barcode对序列的来源进行分类,舍弃无连接的序列。对连接上的序列进行过滤(连续相同碱基<6,模糊碱基N<1),获得最终序列[2]。应用QIIME,根据序列的相似度,将序列归为多个OTU(操作分类单元)[3],同时应用mothur软件中的uchime的方法去嵌合体序列[4]。
1.3 统计学方法 QIIME调用uclust对序列进行聚类,选取每个类最长的序列为代表序列。采用blast的方法对silva数据库进行比对,获得每个OTU分类学信息。OTU产出后,统计各个样品含有OTU情况及每个OTU中含有序列的数目,并采用T-test进行统计学测验发现差异OTU。
2 结果
2.1 阴道微生态状况 观察组中,阴道微生态正常6例(60.00%);阴道微生态失调4例(40.00%)。包括BV患者3例(75.00%);外阴阴道假丝酵母菌病(VVC)患者1例(25.00%)。对照组中,阴道微生态正常7例(70.00%);其余为阴道微生态失调3例(30.00%),包括BV患者2例(66.67%);外阴阴道假丝酵母菌病(VVC)患者1例(33.33%);两组年龄及阴道分泌物pH值比较差异无统计学意义。观察组雌激素均值为99.94pmol/L,对照组雌激素均值为522.84pmol/L,两组比较差异有统计学意义。
2.2 阴道微生态菌群状况 (1) 测序情况统计:本资料对20例样本的16S rDNA进行测序,其中N3和N5两个样本测序失败,未纳入后期分析(见表1)。(2)阴道微生态菌种在产褥期和正常生育期的OTU数目统计:应用QIIME,根据序列的相似度>97%归为一个OTU。结果发现绝大部分的OTU在正常妇女和产褥期妇女中均有检测,占全部OTU的63%。在正常样本中特异性检测到OTU189个,而在产褥期样本中特异性检测到318个。(3)阴道微生态菌种在产褥期和正常生育期的生物多样性分析:在群落丰富度的指数(Chao1指数和ACE指数)的分析发现,两组样本无差异,同时用群落多样性的指数(Shannon指数和Simpson指数)也未发现差异。结果表明无论是产褥期的妇女,还是正常的妇女,其阴道微生态的生物多样性不会有显著性的变化,起变化的可能仅是部分菌在数量上的差异(见表2)。(4)两组样本的主成分分析:采用偏最小二乘方判别分析(PLS-DA)过滤与模型分类不相关信号即正交信号,获得PLSDA模型,其模型质量参数为,R2X=0.423,R2Y=0.9,Q2=0.138。结果显示,正常样本很好的聚为一类,而产褥期的妇女则分为三组,Ⅰ组(T1和T4),Ⅱ组(T2,T5,T6,T7),以及Ⅲ组(T3,T8,T9,T10)。Ⅲ组和正常孕妇聚为一类。(5) 产褥期和正常生育期主要的差异菌:根据PCA的分析结果,为避免干扰,将除Ⅲ组之外的产褥期的样本(即Ⅰ和Ⅱ组)和正常样本进行比较,发现差异的菌属。根据文献报道,健康妇女阴道常驻菌包括乳杆菌(lactobacillus)、假丝酵母菌、B族链球菌(Group B Streptococcus)、表皮葡萄球菌(staphylococcus)、梭状杆菌(Clostridium Septicum)、粪链球菌(Streptococcus faecalis)、消化球菌(peptococcus)、类杆菌(bacteroid),而过路菌主要为金黄色葡萄球菌(Staphylococcus aureus)、肠杆菌(enterobacteria)、丙酸杆菌(Propionibacterium)、消化链球菌(peptostreptococcus)和韦荣球菌(Veillonella)[4]。本资料中两组主要的差异微生物(见表3)。正常妇女的阴道中的乳杆菌属的含量是产褥期妇女的>100倍。进一步分析乳酸杆菌的种类(见表4),此次试验中主要鉴定到的乳杆菌属为Lactobacillus iners和s-未能鉴定的种,这两种菌的含量和在产褥期孕妇中显著降低,另外鉴定到的Lactobacillus brevis和Lactobacillus coleohominis差异不显著,原因可能与其含量较少相关。在产褥期妇女样本中Clostridiales,Peptoniphilus,Acinetobacter等几类菌有显著的升高(见表2)。
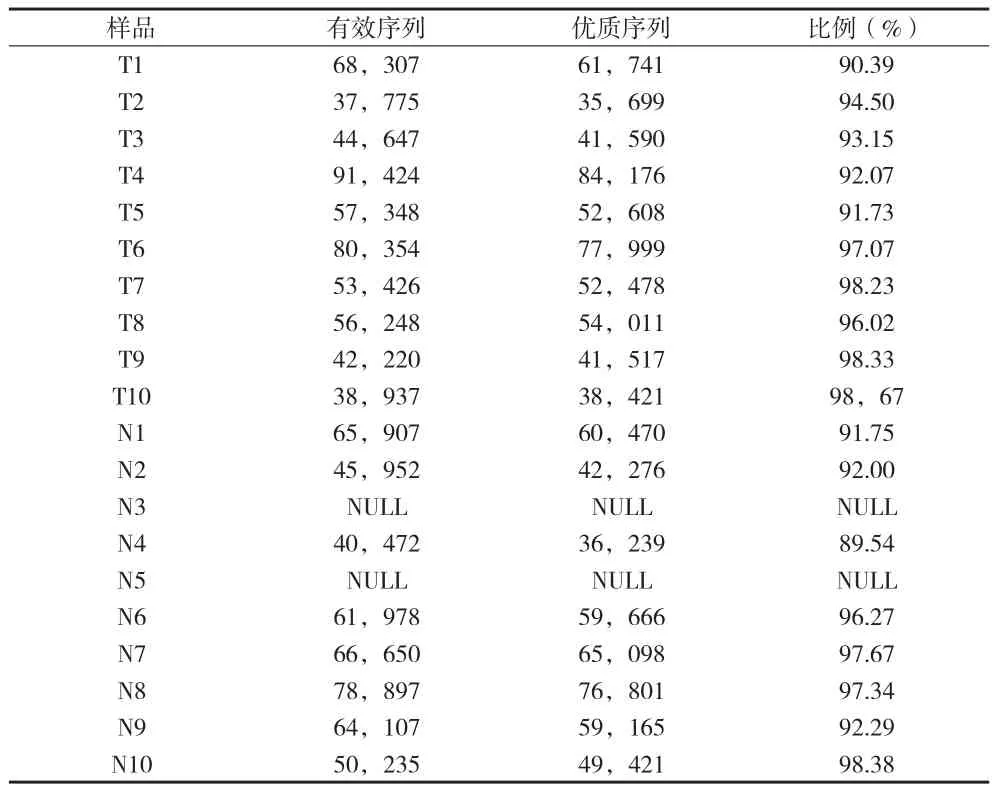
表1 样品序列数统计表
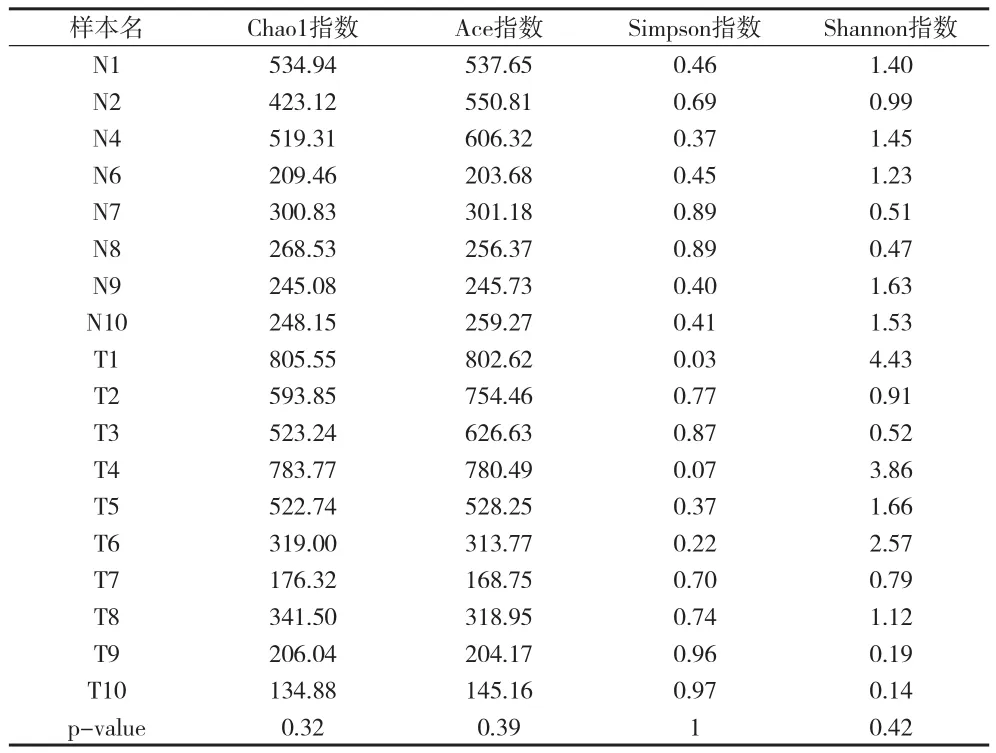
表2 生物多样性指标
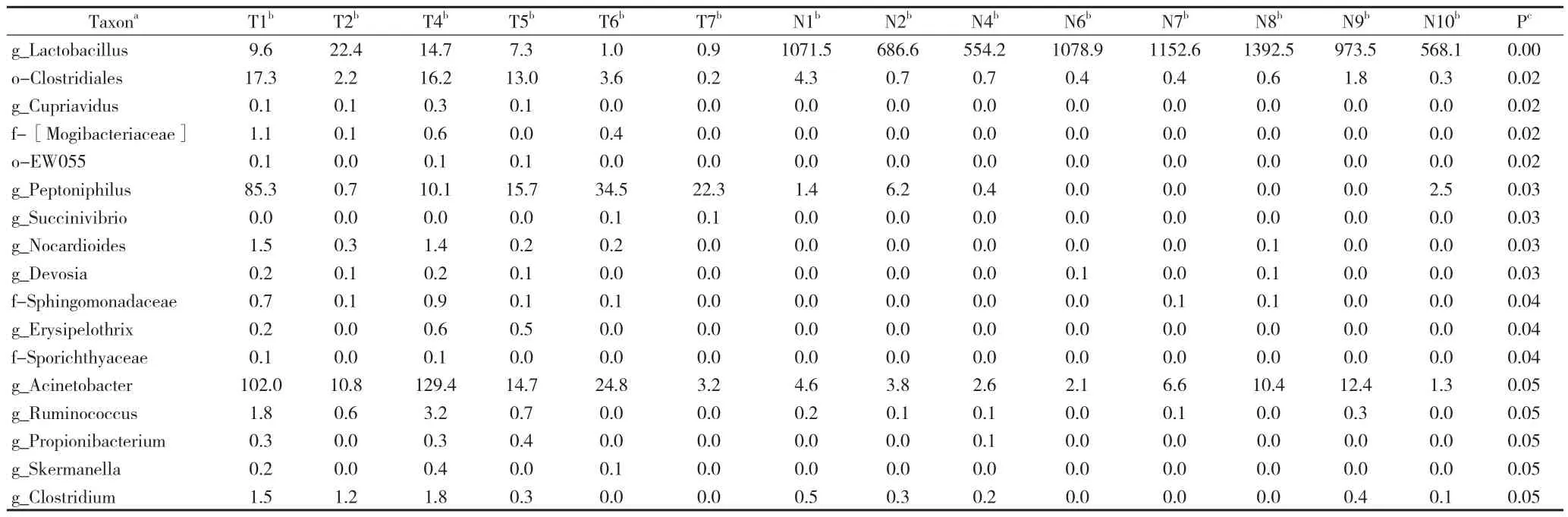
表3 两组妇女阴道中菌属比较

表4 两组妇女的阴道中乳杆菌属比较
3 讨论
正常妇女的微生态一致性较好,基本归为一类,表明在正常妇女的阴道微生态以相当稳定的形式存在。相对于正常孕妇,产褥期孕妇的阴道微生态却以3组形式存在,结合采样时间,作者分析Ⅲ组的采样时间为产褥期的后期,因此,其微生态的结构特征已经倾向于正常妇女,而Ⅰ组和Ⅱ组为相对早期,因此,其微生态与正常妇女有显著差异。表明妇女在产后,其阴道微生态也存在一个逐渐修复的过程,这是一个动态的过程,而这种修复的机制目前尚未明确。
产褥期妇女和正常妇女阴道乳杆菌的差异表明,早期产褥期妇女的阴道微生态有明显的改变。阴道的健康和乳杆菌的数量变化及pH变化均密切相关[5]。乳杆菌是阴道正常菌群中最重要的益生菌成员,其能维持阴道内较低的酸度,从而抑制部分病原菌的生长,维持阴道自净及抗感染中起着关键作用,乳杆菌数量的下降和pH上升会提升早产以及感染性传播疾病的风险。因此,产褥期的妇女尤其要注意避免引起产褥期菌群失调的行为,如产褥期性生活行为、不良阴道清洁方式等[6]。
临床标本中常能分离到的不动杆菌属细菌有醋酸钙不动杆菌、洛菲不动杆菌、溶血不动杆菌、鲍曼不动杆菌、琼氏不动杆菌、约翰逊不动杆菌,最常见的是鲍曼不动杆菌。由不动杆菌引起的医院内感染近10年来增高的趋势明显,且多是多重耐药菌株。最常见的分离部位是呼吸道、尿道和伤口,所致的疾病包括肺炎、心内膜炎、脑膜炎、皮肤和伤口感染、腹膜炎、尿路感染等。在产褥期妇女样本中发现这类菌的水平升高,表明产褥期的妇女的阴道抗菌能力的下降。
综上,通过妇女阴道的细菌菌群的丰度信息可以增进对细菌菌群和健康之间关系的了解。早期只能通过qPCR或者培养对有限的菌群进行相对定量,而现在我们通过16S rDNA测序的方式,可以更为直接的获取相对完整的阴道细菌群落结构的信息并很好的揭示阴道微生态的变化,它有希望成为一个新的、系统的评估手段。不仅可以分析产妇产褥期阴道单一群落恢复情况,而且还能用于检测和评估其综合健康情况。
1 张群英,薛映辛,张艳玲,等.482例产褥期阴道微生态调查.中国微生态学杂志,2011,23:552~553.
2 Ravel J, Gaer P, Abdo Z,et al. Aginal microbiome of reproductive-age women. Proc Natl Acad Sci USA, 2011, 108: 4680~4687.
3 Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27(21):2957~2963.
4 Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 2010,7(5):335~336.
5 Edgar RC, Haas BJ, Clemente JC,et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27(16):2194~2200.
6 李兰娟.感染微生态学.北京:人民出版社,2002:422~424.
Objective The use of a bacterial gene sequencing technology to understand the distribution of vaginal microbial populations. Methods Changxing County randomly selected MCH 42-56 days postpartum women for vaginal secretions (pH value,cleanliness,pus ball,trichomoniasis,mold,etc.) inspection and measurement of estrogen and extracted total microbial genome amplified 16S rRNA sequencing. At the same time,randomly selected control group of normal women of childbearing age,for vaginal secretions (pH value,cleanliness,pus ball,trichomoniasis,mold,etc.) inspection and measurement of estrogen extracted total microbial genome sequencing of 16S rRNA amplification. Compare two Results thus understand postpartum vaginal microfl ora: The species composition and physiological condition. Results By sequencing experiments,we found that in women with normal vagina, the content of Lactobacillus (Lactobacillus) is more than 100 times for puerperal women,at an early maternal,their lactobacilli signifi cantly decreased,which caused the decrease in the protective ability. We also found Acinetobacter species of early maternal vaginal content increased,which also caused the decrease of protective ability. Conclusion Postpartum maternal microfl ora present a dynamic process,early and normal have signifi cant differences in their late microecological consistent with normal women. 16S rRNA sequencing may well reveal changes in vaginal microfl ora,which promises to be a new assessment tools,system ,which can be used not only to analyze the recovery of maternal vaginal microfl ora,but to detect and assess its overall situation.
Vaginal microfl ora 16S rDNA High-throughput sequencing Lactobacillus Acinetobacter.
浙江省医药卫生科技项目(2012KYB202)
313100 浙江省长兴县妇幼保健院 (张群英 王黎 陈祖云)
310012 浙江省微生物研究所(陈欢 方序)
猜你喜欢
中外医学研究(2022年31期)2022-12-30
中国典型病例大全(2022年10期)2022-05-10
中国生殖健康(2020年4期)2021-01-18
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
当代陕西(2019年5期)2019-03-21
中国生殖健康(2018年4期)2018-11-06
中国医药指南(2017年3期)2017-11-13
视野(2017年4期)2017-02-15
中国当代医药(2015年31期)2015-03-01
