
钠离子电池负极材料密度泛函理论研究进展
2016-09-12 06:13:41杨绍斌李思南
功能材料 2016年8期
杨绍斌,李思南,,孙 闻,董 伟,沈 丁
(1. 辽宁工程技术大学 材料科学与工程学院,辽宁 阜新 123000;2. 辽宁工程技术大学 矿业学院,辽宁 阜新 123000)
钠离子电池负极材料密度泛函理论研究进展
杨绍斌1,李思南1,2,孙闻1,董伟2,沈丁1
(1. 辽宁工程技术大学 材料科学与工程学院,辽宁 阜新 123000;2. 辽宁工程技术大学 矿业学院,辽宁 阜新 123000)
近年来有关钠离子电池的理论计算和实验研究不断增多,其中第一性原理计算的应用及结果引起国内外学者的研究兴趣。综述了密度泛函理论在钠离子电池负极材料中应用的研究进展,主要包括结构模型、电极电势或电池电压、可逆容量、晶格膨胀及其弹性、钠的存在状态、钠离子的扩散行为和电子导电性。随着研究的逐步深入,通过密度泛函理论计算能够给出电极材料可逆储钠性能的系统解释,并在新材料结构和成分设计方面发挥更重要的作用。
钠离子电池;负极材料;第一性原理;密度泛函理论
0 引 言
钠离子电池与成熟的锂离子电池技术相比仍处于研发阶段,而钠资源丰富,易于开采,钠离子电池成本更低,在大型电能储存设备中更具市场优势[1-2]。近几年,钠离子电池电极材料的实验及计算研究不断增多,获得了众多满意结果,同时,可逆容量低和循环稳定性差等问题也逐渐显露出来。为获得比容量高、电池稳定性良好的钠离子电池电极材料,分析充放电过程中钠离子的嵌入脱出机制、电极材料的结构及电化学性能等显得尤为重要。密度泛函理论计算无疑是一种有效的分析方法,已被广泛用于材料的电子结构和物理化学性质的研究,并在锂离子电池电极材料研究领域得到广泛应用[3-4],而在钠离子电池电极材料的研究中正极材料居多,近年来负极材料的研究刚刚引起国内外学者的关注,并迅速发展,主要涉及第Ⅳ主族、第Ⅴ主族(除氮)和无机化合物材料,研究内容关于晶体结构、电极电势或电池电压、可逆容量、钠离子的扩散行为和电子导电性、结构弹性等。相信随着钠离子电池负极材料研究逐步深入,密度泛函理论将会在新型电极材料的设计研发,以及对实验现象的理论解释方面发挥更重要作用。因此,本文综述了近年来第一性原理在钠离子电池负极材料中应用的研究进展。
1 能量计算与结构
1.1碳基负极材料
石墨负极材料已经在锂离子电池中获得大规模应用,而实验和理论研究表明钠离子难以嵌入石墨层间[5-7],限制了石墨在钠离子电池中的应用。通过第一性原理计算能够解释Na的嵌入脱出机制,进而找出改善石墨储钠性能的途径。
Kunihiro等[8]采用PBE(perdew-burke-ernzerh)形式的广义梯度近似(generalizedgradientapproximation,GGA[9]),计算了碱金属石墨嵌入化合物(AM-GICs,见图1)形成能ΔE与嵌入浓度之间的关系,与Li和K相比Na-GIC热力学不稳定,同时,计算得到Na-GIC中钠原子引起的结构畸变较大,对石墨片层产生挤压,阻碍钠原子嵌入。对于弱相互作用体系,传统的密度泛函方法GGA和局域密度近似(localdensityapproximation,LDA)交换关联函数不适合处理色散力,需要引入范德华(vdW)修正处理交换关联势,vdW泛函修正形式较多,而通过一种泛函很难同时实现结构和能量的准确预测,同时不同泛函应用于同一体系的能量计算又会引入新的误差,因此,在已知众多方法中选择合适的泛函尤为重要。Okamoto等[6]认为采用vdW修正的交换关联函数vdW-DF-C09和vdW-DF2-C09适合预测石墨的片层间距;而vdW-DF和vdW-DF2适合预测一阶GICs的结构;vdW-DF-C09泛函低估石墨片层结合能。
Tsai等[10]测试了9种vdW泛函, 其中optPBE-vdW方法能够较好的预测石墨和Na-GIC的结构参数,低嵌入浓度时纯石墨和无定形碳中3种缺陷结构的嵌入电势与片层间距的关系如图2,发现石墨层间距>0.394nm时钠离子能够嵌入,大于实验值(0.37~0.38nm),表明无定型碳中的缺陷降低了层间距的限制,其中,MV型缺陷最利于钠离子的嵌入。

图1 碱金属石墨嵌入化合物模型[8]

图2 低嵌入浓度下纯石墨烯、MV、DV和SW层间距与嵌入电势关系[10]
Fig2ThesodiationpotentialatlowNacontentsasafunctionoftheinitialinterlayerdistancesforpristine,MV,DVandSW[10]
一般认为,碱金属吸附在石墨表面的六边形中心上方[11-13],碱金属吸附能Li(1.21eV)>Cs(1.04eV)≥Rb(1.02eV)≥K(0.99eV)>Na(0.55eV),Na与石墨的结合力最弱。Rytkönen等[13]通过GGA-PBE方法发现Na原子在石墨表面容易形成Na3、Na4、Na5型团簇结构,与实验观察到的岛状结构相似。
石墨烯是一种呈2D蜂窝状结构的新型纳米材料,是单层、双层和少层(3~10层)石墨烯的统称[14-15]。由于其特殊的电化学性能,近年来,已有众多学者[16-24]将石墨烯应用到锂和钠离子电池研究中。DFT计算表明碱金属原子最可能吸附在石墨烯表面的六边形中心上方[18-24],但有少数研究[25]将碱金属作为“类氢原子”(吸附在C原子上方附近)处理。通常采用不同软件和计算方法获得的吸附能和吸附高度略有差异,见表1所示,但结果均表明Na与石墨烯表面结合力最弱。

表1 碱金属的吸附能及吸附高度
Chen等[26]指出Na在石墨烯表面吸附热力学不稳定,通过掺杂硼能够改善石墨烯吸附能力,计算得到硼掺杂石墨烯BC3理论容量达762mAh/g。Malyi等[27]指出钠离子能够吸附在石墨烯的位缺陷附近,缺陷能够增强对Na的吸附能力。

图3 硼掺杂石墨烯的晶体结构[26]
实验发现钠在石墨烯纳米片中具有较高的比容量和较大不可逆容量[28]。而实际制备的石墨烯含有一定量的缺陷、微孔及O和H原子,已有的理论预测结果表明缺陷和适当的掺杂能够提高石墨烯储钠能力,计算研究具有不同尺度和浓度缺陷的、不同层数和掺杂形式的石墨烯储钠过程,将成为今后石墨烯储钠性能的研究方向。
1.2第Ⅳ主族材料
Na在Si中主要以间隙缺陷存在,计算表明Na在无定型Si中形成能(0.09eV)小于晶体Si(1.51eV),Na更容易嵌入无定型Si,同时硅的无定型化能够降低体积膨胀率[29]。Kulish等[30]采用GGA-PBE方法,系统研究了Na在单质Si、硅烷聚合物(polysilane)和氢化硅烯(silicene)中的嵌入过程。发现Na嵌入层状polysilane和silicene中的形成能分别为-0.57和-0.32eV,比单质硅(+0.60eV)小,钠原子更易于嵌入polysilane和silicene中。
Yue等[31]通过自组装反应合成了一种由无定型Ge镀层覆盖的纳米棒状阵列结构材料Si/TiN/Ti(见图4),如图4(b),由于Na在Ge(100)晶胞中的亚稳态能量差(3.18eV)比Si(100)晶胞(2.14eV)大,表明Na原子更容易在Ge结构中找到稳定位置。

图4 Si/TiN/Ti/Ge纳米棒状阵列的自组装过程[31]
对于第Ⅳ主族单质材料,无定型结构能够提高储钠能力和结构稳定性。计算方面,通常建立无定型模型需要包含大量原子,但因对称性差计算耗时较长,且模型结构缺乏实验支撑,给无定型结构理论模型建立带来困难。
1.3第Ⅴ主族材料
关于Na-P体系的实验研究较多[2,32],利用第一性原理计算的相关研究较少。Yu等[33]研究了Na3P中离子和电子传导性能。如图5所示,Na3P是由含P层和Na原子层交替堆垛而成,Naeq、Naax分别表示含P层和不含P层中的Na原子。
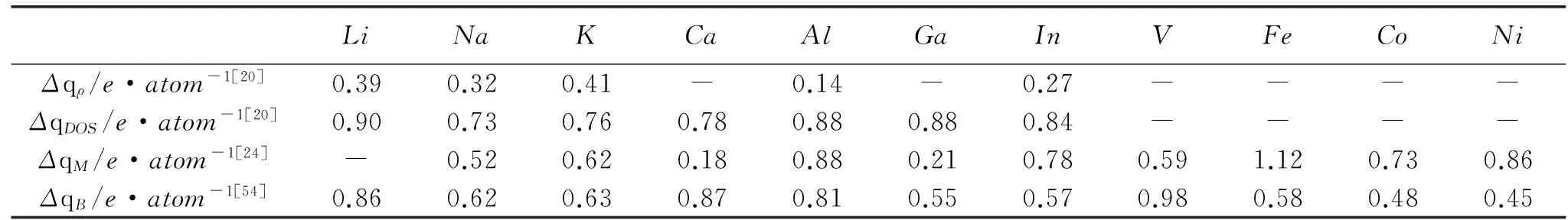
Mortazavi等[34]利用DFT研究了P、As、Sb、Bi储钠能力,发现钠离子的嵌入导致体积膨胀40%~400%(Bi 与第Ⅳ主族材料相似,2D结构P和无定型P具有较好的循环稳定性[37,39]而有关Na离子在第Ⅴ主族的无定型材料中嵌入/脱出过程,以及无定型结构储钠机制和性能预测,有待进一步研究。 图5 Na3P的晶体结构及钠离子扩散路径[33] 1.4化合物 层状化合物是钠离子电池负极材料研究的重点。化合物的形成能或吸附能计算通常采用GGA-PBE方法,层状零应变材料P2-Na0.66[Li0.22Ti0.78]O2结构稳定具有极好的循环性能[40],通过DFT计算得到体积变化率仅为0.21%。Dequan等[41]通过第一性原理计算预测了Li、Na、K和Ca在单层Ti3C2表面吸附位置,吸附能的计算表明Na比Li更容易吸附在单层Ti3C2表面,且双边吸附的最大容量(351.8mAh/g)是单边吸附的两倍。Mortazavi等[42]系统研究了Na在MoS2中的嵌入过程,发现NaxMoS2中Na含量增至x=0.39,引起2H-MoS2→1T-MoS2相变,当0≤x≤1时,MoS2热力学稳定。Su等[43]计算发现单层1H-MoS2双面吸附最大理论容量为335mAh/g是体结构2H-MoS2理论容量的2倍。Legrain等[44]利用DFT方法对比了Na在锐钛矿、金红石、TiO2-B和无定形结构的TiO2中的形成能,结果表明无定形TiO2储钠能力更优异。 嵌入电压是钠离子电池设计的一个重要参数,一般负极材料的电压平台要求足够低,以获得工作电压较高的钠离子电池,同时要保证电解液的稳定。平均嵌入电压可根据电极反应式,通过计算材料基态总能量获得。 Chevrier等[45]计算了Na在Si、Ge、Sn和Pb的平均电压曲线(见图6),以Si为例,平均电压V的计算方法如下: 电极反应方程式 其中,E表示通过DFT计算得到的相应结构总能量,z=1,为Na所带电荷数,e表示电子电量的绝对值。由图6可以知,Na在第Ⅳ主族单质Si、Ge、Sn和Pb中的平均电压<1 V,Si、Ge、Sn和Pb满足实际电池电极电势的应用要求。Fleur等[29]采用PBE泛函和DZP基组,得到Na原子在a-Si中的平均嵌入电压较小,仅为0.14 V。Mortazavi等[34]采用自旋极化密度泛函方法计算了钠离子嵌入单质P、As、Sb、Bi的电极电势(即平均电压,如图7所示),计算结果与实验值符合较好。 Wang等[40]通过DFT计算得到P2-Na0.66[Li0.22Ti0.78]O2的平均嵌入电压为0.47 V,比实验值(0.75 V)略低,进一步的实验表明实际成分为Na0.63[Li0.22Ti0.78]O1.98,实际材料中存在Na和O空位,导致这种差异。Su等[43]计算了单层MoS2的电压平台约为1.0 V,体结构MoS2为1.7~2.0 V(0.75~1.25 V[42]),因此单层MoS2具有较高的全电池电压。 f[45-46]能够反应充放电过程体积膨胀率与容量的关系,计算方法如下 其中,F表示法拉第常数,F=26.802Ah/mol;ξf为相对体积膨胀率;Vavg为全电池平均电压(正极电压取3.75V);υ表示每摩尔金属原子所占据的体积。 Chevrier等[45]给出了Na嵌入Si、Ge、Sn、Pb和硬碳中的体积能量密度,当负极材料在粘结剂作用下承受100%的体积膨胀率时,Na-Si合金的体积能量密度最大,约为2.7Wh/cc。Mortazavi等[34]利用DFT研究了P、As、Sb、Bi中不同钠离子嵌入浓度的体积能量密度,体积膨胀率为100%时,Na-Bi合金的体积能量密度最大,约为2.4Wh/cc。而Li-Si合金的体积能量密度较高(100%体积膨胀率时约为4.8Wh/cc[29]),这表明若不考虑成本,当体积膨胀率或容量一定时,锂离子电池优于钠离子电池。 图6 NaxM的电极电势[45] 图7 DFT计算Na-M电极电势[34] DFT计算不仅广泛用于能量计算和结构优化,还是离子传导机制研究的有效途径。Wan等[47]采用GGA-PBE方法计算得到高阶碱金属石墨嵌入化合物中Na的扩散能垒为0.72eV,高于Li(0.64eV)和K(0.53eV)。Wang等[48]采用optPBE-vdW范德瓦尔斯修正方法,对Li、Na、K在石墨中的一阶嵌入化合物的扩散行为进行了研究,计算得到一阶嵌入化合物NaC6中的扩散激活能为0.28eV,比LiC6(0.47eV)小得多。 Rytkönen等[49]研究发现的碱金属在HOPG表面的吸附能,Li(0.21eV)>Na(0.06eV)>K(0.05eV)>Rb(0.03eV)>Cs(0.02eV),原子半径越大越容易扩散。Wehling等[21]得到碱金属在单层石墨烯表面和石墨表面的扩散能垒具有相同排序规律,Li(0.31eV)>Na(0.09eV)>K(0.06eV)>Cs(0.04eV)。Nakada等[19]采用投影缀加平面波(PAW)赝势,计算了元素周期表中几乎所有原子在单层石墨烯表面的吸附能和扩散能垒(见图8),同时,定义了原子迁移的“门槛能”约为0.5eV,即扩散能垒<0.5eV时,室温下原子容易迁移。Na原子的扩散能垒仅为0.13eV(0.09eV[21]),因此,室温下Na原子在石墨烯表面容易迁移。Chen等[26]计算了硼掺杂石墨烯吸附Na的扩散能垒0.16~0.28eV,与石墨烯相近。 图8 不同原子最稳定位置扩散能垒[19] Malyi等[50-51]采用DFT方法计算了低嵌入浓度时Na在Si中的扩散能垒达1.14eV,通过掺Na使得扩散能垒降至0.28eV,可以推测,通过控制钠离子掺杂浓度能够提高电池的充放电速率。Yue等[31]通过第一性原理计算得到Na在Ge(100)晶胞中扩散能垒(4.01eV)小于Si(100)晶胞中扩散能垒(5.89eV),表明Na更容易在Ge电极中扩散。Kulish等[30]采用NEB方法给出了Na在体Si、Polysilane和Silicene中的扩散过程(见图9),Na在层状结构Polysilane(0.41eV)和Silicene(0.12eV)中的扩散能垒比体结构Si(1.06eV)小得多。 图9 Na的嵌入位置和扩散路径 Yu等[33]给出了Na3P中可能扩散路径Ⅰ、Ⅱ和路径ⅢI逆向的扩散能垒分别为0.038,0.040和0.018eV,见图5(c)。Liu等[38]计算得到Na吸附浓度低时,在磷烯表面具有较低扩散能垒0.040~0.063eV(0.040~0.380eV[37]),高吸附浓度时仍小于0.070eV。 Mortazavi等[42]计算得到Na在体结构2H-MoS2和1T-MoS2中扩散能垒分别为0.68和0.28eV,在1T-MoS2中更有利于离子传导。Su等[43]给出Na在单层MoS2中的扩散能垒为0.11eV(0.086eV[52]),小于体结构的扩散能垒,钠离子更容易在单层MoS2中扩散,扩散路径见图10所示。 Chen等[53]构建了TiO2/graphene复合物模型,图11为O—C原子部分结合模型中钠离子扩散路径,为避免GGA+U高估碱金属的电荷转移势垒,采用GGA-PBE方法,计算得到复合物模型中扩散能垒(0.2eV)小于体结构TiO2-B(2.2eV)。 综上所述,层状结构材料具有较低的扩散能垒,其中单层结构扩散能垒最低。对于不同结构类型的材料,还未找到一种普适规则确定扩散路径。充放电过程中钠离子溶度影响离子迁移速率,而有关Na离子间相互作用机制的研究较少,还需进行深入理论研究。石墨烯、MoS2和Ti3C2等单层负极材料的表面扩散能垒较低,有利于离子传导,但是单层石墨烯吸附能力较弱,同时比表面积大,易产生较大的不可逆容量,将是单层MoS2和Ti3C2负极材料亟待解决的问题。 图10 Na在体2H-MoS2层间和单层MoS2表面的扩散路径[43] 图11 Na在石墨烯与TiO2-B(001)面部分结合模型中的扩散[53] 钠离子电池研究中,电子转移数通常由以下几种方法获得[11,20,54]:(1) 通过对电荷密度积分来测定(Δqρ);(2) 通过态密度(densityofstate,DOS)中获得的费米能级之差确定(ΔqDOS);(3)Mulliken布局分析(ΔqM);(4)Bader布局分析(ΔqB)。Caragiu[11]利用GGA和局域自旋密度近似(localspindensityapproximation,LSDA)方法,通过DOS计算得到Na原子在石墨表面吸附的电子转移数<0.2e/atom,并且吸附浓度影响小,认为Na与石墨表面成非离子键,与Johnson等的结果一致[55]。Chan等[20]发现DOS方法适合处理石墨烯吸附碱金属的离子键模型,而石墨烯与过渡族金属间形成的共价键强烈影响DOS结构,DOS方法将不适用。准原子基最小轨道法(QUAMBOs)[24]则能够处理由离子键和共价键结合的系统,利用QUAMBOs,Mulliken电荷布局分析可以经过VASP波函数计算的后处理获得,弥补了密度泛函理论做Mulliken布局分析的不足。上述方法计算得到的金属原子在石墨烯表面吸附的电子转移数列于表2,结果虽然有差别,但碱金属中Na的电子转移数最少,Na的离子性最弱。 Dequan等[41]通过基于DFT的Bader电荷布局分析,给出了Na在单层Ti3C2表面电荷转移数分别为0.40e/atom大于Li(0.21e/atom),Na与表面结合更强。 表2 吸附原子与石墨烯间的电子转移数 Chen等[26]发现钠在硼掺杂石墨烯表面吸附使带隙消失,从半导体转变为金属态,有利于电子传导。Su等[56]利用ABINIT软件包,采用LDA方法和超软赝势,计算发现Na3Bi中Na和Bi的DOS曲线产生叠加,指出Bi能够为钠离子的扩散和驻留提供足够空间。 本文介绍的钠离子电池负极材料的晶体结构实验参数、计算方法、实验和理论容量列于表3。 表3 钠离子电池负极材料结构参数、计算方法及比容量 详细综述了钠离子电池的第Ⅳ主族、第Ⅴ主族(除氮)和化合物负极材料第一性原理的研究进展,相关研究主要包括如下几个方面:建立和优化晶体结构、计算电极电势或电池电压、可逆容量(钠离子吸附或嵌入形成能及结构缺陷影响)、分析结构稳定性(体积膨胀率)、充放电速率(钠离子扩散能垒和路径)、材料导电性(态密度)。 石墨负极材料在锂离子电池中得到了广泛应用,由于钠的石墨嵌入化合物热力学不稳定以及层间扩散能垒较高,储钠能力受到限制,增加层间距和引入结构缺陷可以降低形成能从而提高储钠能力,因此无定形碳仍然是人们研究的重要方向。体积膨胀率小的含钛化合物负极材料具有良好的循环性能,寻找零应变电极材料仍然是人们努力的方向。而第Ⅳ和Ⅴ主族单质(除C和N)由于体积膨胀较大,借鉴锂离子电池负极材料研究经验,很难走入实用化阶段。层状结构材料由于其特殊的表面和界面效应,通常表现出优良的吸附性能和离子传导性能,将成为未来电极材料研究的重点;石墨烯材料将继续是钠离子电池领域研究热点,以后的研究将更倾向于通过密度泛函理论预测石墨烯复合材料储钠能力。随着DFT在钠离子电池电极材料领域研究的逐步深入和计算能力的不断提升,将使研发成本降低,研发速度进一步提高。 [1]SlaterMD,KimD,LeeE,etal.Sodium-ionbatteries[J].AdvancedFunctionalMaterials, 2013, 23(8): 947-958. [2]HeHN,WangHY,TangYG,etal.Currentstudiesofanodematerialsforsodiumionbattery[J].ProgressinChemistry, 2014, 26(4): 572-581. 何菡娜, 王海燕, 唐有根, 等. 钠离子电池负极材料[J].化学进展, 2014, 26(04): 572-581. [3]OngSP,ChevrierVL,HautierG,etal.Voltage,stabilityanddiffusionbarrierdifferencesbetweensodium-ionandlithium-ionintercalationmaterials[J].Energy&EnvironmentalScience, 2011, 4(9): 3680-3688. [4]YangSB,LiB,TangSW.Reviewoffirstprinciplecalculationsofanodematerialsinlithiumionbatteries[J].MaterialsReview, 2012, 26(8):62-66. 杨绍斌, 李犇, 唐树伟. 第一性原理在锂离子电池负极材料方面的应用研究[J].材料导报, 2012, 26(15): 62-66. [5]AshenRC,WilsonSA.Lamellarcompoundofsodiumwithgraphite[J].Nature, 1958, 181(4606): 409-410. [6]OkamotoY.Densityfunctionaltheorycalculationsofalkalimetal(Li,Na,andK)graphiteintercalationcompounds[J].TheJournalofPhysicalChemistryC, 2014, 118:16-19. [7]GeP,FouletierM.Electrochemicalintercalationofsodiumingraphite[J].SolidStateIonics, 1988, 28: 1172-1175. [8]NobuharaK,NakayamaH,NoseM,etal.First-principlesstudyofalkalimetal-graphiteintercalationcompounds[J].JournalofPowerSources, 2013, 243: 585-587. [9]PerdewJP,BurkeK,ErnzerhofM.Generalizedgradientapproximationmadesimple[J].PhysicalReviewLetters, 1996, 77(18): 3865. [10]TsaiP,ChungSC,LinS,etal.Abinitiostudyofsodiumintercalationintodisorderedcarbon[J].JournalofMaterialsChemistryA, 2015, 3(18): 9763-9768. [11]CaragiuM,FinbergS.Alkalimetaladsorptionongraphite:areview[J].JPhys:CondensMatter, 2005, 17:R995-R1024. [12]ZhuZH,LuGQ.ComparativestudyofLi,NaandKadsorptionsongraphitebyusingabinitiomethod[J].Langmuir, 2004, 20(24): 10751-10755. [13]RytkönenK,AkolaJ,ManninenM.Sodiumatomsandclustersongraphitebydensityfunctionaltheory[J].PhysRevB, 2004, 69(20): 205404. [14]NovoselovKS,GeimAK,MorozovSV,etal.Electricfieldeffectinatomicallythincarbonfilms[J].Science, 2004, 306(5696): 666-669. [15]RaoCNR,SoodAK,SubrahmanyamKS,etal.Graphene:thenewtwo-dimensionalnanomaterial[J].AngewandteChemieInternationalEdition, 2009, 48(42): 7752-7777. [16]WangGX,ShenXP,YaoJ,etal.Graphenenanosheetsforenhancedlithiumstorageinlithiumionbatteries[J].Carbon, 2009, 47(8): 2049-2053. [17]SuD,AhnHJ,WangG.SnO2@graphenenanocompositesasanodematerialsforNa-ionbatterieswithsuperiorelectrochemicalperformance[J].ChemCommun, 2013, 49(30): 3131-3133. [18]QiaoL,QuCQ,ZhangHZ,etal.Effectsofalkalimetaladsorptiononthestructuralandfieldemissionpropertiesofgraphene[J].DiamondandRelatedMaterials, 2010, 19(11): 1377-1381. [19]NakadaK,IshiiA.MigrationofadatomadsorptionongrapheneusingDFTcalculation[J].SolidStateCommunications, 2011, 151(1): 13-16. [20]KevinTChan,JBNeaton,MarvinLCohen.First-principlesstudyofmetaladatomadsorptionongraphene[J].PhysicalReviewB, 2008, 77:235430-1-235430-12. [21]WehlingTO,KatsnelsonMI,LichtensteinAI.Impuritiesongraphene:midgapstatesandmigrationbarriers[J].PhysicalReviewB, 2009, 80(8): 085428. [22]KlintenbergM,LebegueS,KatsnelsonMI,etal.TheoreticalanalysisofthechemicalbondingandelectronicstructureofgrapheneinteractingwithgroupIAandgroupVIIAelements[J].PhysicalReviewB, 2010, 81(8): 085433. [23]JinKH,ChoiSM,JhiSH.Crossoverintheadsorptionpropertiesofalkalimetalsongraphene[J].PhysicalReviewB, 2010, 82(3): 033414. [24]LiuXJ,WangCZ,YaoYX,etal.Bondingandchargetransferbymetaladatomadsorptionongraphene[J].PhysicalReviewB, 2011, 83(23): 235411. [25]MedeirosPVC,deBritoMotaF,MascarenhasAJS,etal.Adsorptionofmonovalentmetalatomsongraphene:atheoreticalapproach[J].Nanotechnology, 2010, 21(11): 115701. [26]LingC,MizunoF.Boron-dopedgrapheneasapromisinganodeforNa-ionbatteries[J].PhysicalChemistryChemicalPhysics, 2014, 16(22): 10419-10424. [27]MalyiOI,SopihaK,KulishVV,etal.AcomputationalstudyofNabehaviorongraphene[J].AppliedSurfaceScience, 2015, 333: 235-243. [28]SuD,AhnHJ,WangG.SnO2@graphenenanocompositesasanodematerialsforNa-ionbatterieswithsuperiorelectrochemicalperformance[J].ChemCommun, 2013, 49(30): 3131-3133. [29]LegrainF,MalyiOI,ManzhosS.ComparativecomputationalstudyoftheenergeticsofLi,Na,andMgstorageinamorphousandcrystallinesilicon[J].ComputationalMaterialsScience, 2014, 94: 214-217. [30]KulishVV,MalyiOI,NgMF,etal.ControllingNadiffusionbyrationaldesignofSi-basedlayeredarchitectures[J].PhysicalChemistryChemicalPhysics, 2014, 16(9): 4260-4267. [31]YueC,YuY,SunS,etal.HighPerformance3DSi/GenanorodsarrayanodebufferedbyTiN/Tiinterlayerforsodium-ionbatteries[J].AdvancedFunctionalMaterials, 2015, 25(9): 1386-1392. [32]SangsterJM.Na-P(sodium-phosphorus)system[J].JournalofPhaseEquilibriaandDiffusion, 2010, 31(1): 62-67. [33]YuXF,GiacomoG,UshiyamaH,etal.First-principlesstudyoffastNadiffusioninNa3P[J].ChemicalPhysicsLetters, 2004, 612(18): 129-133. [34]MortazaviM,YeQJ,BirbilisN,etal.Highcapacitygroup-15alloyanodesforNa-ionbatteries:electrochemicalandmechanicalinsights[J].JournalofPowerSources, 2015, 285: 29-36. [35]LiuH,NealAT,ZhuZ,etal.Phosphorene:anunexplored2Dsemiconductorwithahighholemobility[J].ACSNano, 2014, 8(4): 4033-4041. [36]KulishVV,MalyiOI,PerssonC,etal.Adsorptionofmetaladatomsonsingle-layerphosphorene[J].PhysicalChemistryChemicalPhysics, 2015, 17(2): 992-1000. [37]KulishVV,MalyiOI,PerssonC,etal.PhosphoreneasananodematerialforNa-ionbatteries:afirst-principlesstudy[J].PhysicalChemistryChemicalPhysics, 2015, 17(21): 13921-13928. [38]LiuX,WenY,ChenZ,etal.Afirst-principlesstudyofsodiumadsorptionanddiffusiononphosphorene[J].PhysicalChemistryChemicalPhysics, 2015, 17(25): 16398-16404. [39]QianJ,WuX,CaoY,etal.Highcapacityandratecapabilityofamorphousphosphorusforsodiumionbatteries[J].AngewandteChemie, 2013, 125(17): 4731-4734. [40]WangYS,YuXQ,XuSY,etal.Azero-strainlayeredmetaloxideasthenegativeelectrodeforlong-lifesodium-ionbatteries[J].NatureCommunications, 2013, 3365. [41]ErD,LiJW,NaguibM,etal.Ti3C2MXeneasahighcapacityelectrodematerialformetal(Li,Na,K,Ca)ionbatteries[J].ACSAppliedMaterialsandInterfaces, 2014, 6:11173-11179. [42]MortazaviM,WangC,DengJK,etal.AbinitiocharacterizationoflayeredMoS2asanodeforsodium-ionbatteries[J].JournalofPowerSources, 2014, 268: 279-286. [43]SuJC,PeiY,YangZH,etal.Abinitiostudyofgraphene-likemonolayermolybdenumdisulfideasapromisinganodematerialforrechargeablesodiumionbatteries[J].RSCAdvances, 2014, 4(81): 43183-43188. [44]LegrainF,MalyiO,ManzhosS.Insertionenergeticsoflithium,sodium,andmagnesiumincrystallineandamorphoustitaniumdioxide:Acomparativefirst-principlesstudy[J].JournalofPowerSources, 2015, 278: 197-202. [45]ChevrierVL,CederG.ChallengesforNa-ionnegativeelectrodes[J].JournaloftheElectrochemicalSociety, 2011, 158(9):A1011-A1014. [46]ObrovacMN,ChristensenL,LeDB,etal.Alloydesignforlithium-ionbatteryanodes[J].JournalofTheElectrochemicalSociety, 2007, 154(9):A849-A855. [47]WanW,WangH.Studyonthefirst-principlescalculationsofgraphiteintercalatedbyalkalimetal(Li,Na,K)[J].IntJElectrochemSci, 2015, 10: 3177-3184. [48]WangZ,RatvikAP,GrandeT,etal.DiffusionofalkalimetalsinthefirststagegraphiteintercalationcompoundsbyvdW-DFTcalculations[J].RSCAdvances, 2015, 5(21): 15985-15992. [49]RytkönenK,AkolaJ,ManninenM.Densityfunctionalstudyofalkali-metalatomsandmonolayersongraphite(0001) [J].PhysicalReviewB, 2007, 75(7): 075401. [50]MalyiOI,TanTL,ManzhosS.Acomparativecomputationalstudyofstructures,diffusion,anddopantinteractionsbetweenLiandNainsertionintoSi[J].AppliedPhysicsExpress, 2013, 6(2): 027301. [51]MalyiOI,KulishVV,TanTL,etal.AcomputationalstudyoftheinsertionofLi,Na,andMgatomsintoSi(111)nanosheets[J].NanoEnergy, 2013, 2(6): 1149-1157. [52]XieXQ,AoZM,SuDW,etal.MoS2/graphenecompositeanodeswithenhancedperformanceforsodium-ionbatteries:theroleofthetwo-dimensionalheterointerface[J].AdvancedFunctionalMaterials, 2015, 25(9): 1393-1403. [53]ChenCJ,WenYW,HuXL,etal.Na+intercalationpseudocapacitanceingraphene-coupledtitaniumoxideenablingultra-fastsodiumstorageandlong-termcycling[J].NatureCommunications, 2015:1-6. [54]NakadaK,IshiiA.DFTcalculationforadatomadsorptionongraphene[M].INTECHOpenAccessPublisher, 2011. [55]JohnsonMT,StarnbergHI,HughesHP.Electronicstructureofalkalimetaloverlayersongraphite[J].SurfaceScience, 1986, 178: 290-299. [56]SuDW,DouSX,WangGX.Bismuth:anewanodefortheNa-ionbattery[J].NanoEnergy, 2015, 12: 88-95. [57]EllisLD,WilkesBN,HatchardTD,etal.InSituXRDstudyofsilicon,leadandbismuthnegativeelectrodesinnonaqueoussodiumcells[J].JournalofTheElectrochemicalSociety, 2014, 161(3):A416-A421. [58]TranTT,ObrovacMN.Alloynegativeelectrodesforhighenergydensitymetal-ioncells[J].JournalofTheElectrochemicalSociety, 2011, 158(12):A1411-A1416. [59]KomabaS,MatsuuraY,IshikawaT,etal.RedoxreactionofSn-polyacrylateelectrodesinaproticNacell[J].ElectrochemistryCommunications, 2012, 21: 65-68. [60]MortazaviM,DengJ,ShenoyVB,etal.ElasticsofteningofalloynegativeelectrodesforNa-ionbatteries[J].JournalofPowerSources, 2013, 225: 207-214. [61]DarwicheA,MarinoC,SougratiMT,etal.BettercyclingperformancesofbulkSbinNa-ionbatteriescomparedtoLi-ionsystems:Anunexpectedelectrochemicalmechanism[J].JournaloftheAmericanChemicalSociety, 2012, 134(51): 20805-20811. [62]FehseM,VentosaE.IsTiO2(B)thefutureoftitanium-basedbatterymaterials[J].ChemPlusChem, 2015, 80(5): 785-795. Density functional theory of anode in sodium-ion batteries YANG Shaobin1,LI Sinan1,2,SUN Wen1,DONG Wei2,SHEN Ding1 (1.Materials Science and Engineering,Liaoning Technical University, Fuxin 123000, China;2.College of Mining, Liaoning Technical University, Fuxin 123000, China) In recent years, increasing researches of theoretical calculation and experimental about the sodium ion battery, the application of the first principle calculation and the results have attracted enormous interest of researchers. The density functional theory of anode materials in sodium-ion batteries has been reviewed in this paper, mainly including structure models of the anode materials, electrode potential or average voltage, reversible capacity, structure expansion and elasticity, existence state of sodium, behavior of ion diffusion and electronic conductivity. With the development of the density functional theory, a systematic explanation of reversible sodium storage behavior of the electrode materials will be given and a significant role will be played in design of structure and composition for the new materials. sodium-ion battery;anode material;first principle;density functional theory 1001-9731(2016)08-08020-11 国家自然科学基金重点资助项目(51274119) 2015-09-10 2015-12-15 通讯作者:杨绍斌,E-mail:lgdysb@163.com 杨绍斌(1963-),男,辽宁阜新人,教授,从事锂、钠离子电池储能材料、碳素功能材料研究。 O646 ADOI:10.3969/j.issn.1001-9731.2016.08.004
2 电压和体积能量密度


3 扩散能垒与扩散路径




4 态密度与电荷转移

5 结 语
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
分子催化(2022年1期)2022-11-02 07:10:16
仪器仪表用户(2021年10期)2021-11-27 08:26:14
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
电子制作(2019年15期)2019-08-27 01:12:04
测控技术(2018年9期)2018-11-25 07:44:44
上海大中型电机(2017年4期)2017-02-06 05:26:59
中华老年多器官疾病杂志(2016年2期)2016-01-16 03:15:46
电源技术(2015年2期)2015-08-22 11:28:30
