
利用RNAi抑制cre1基因转录提高里氏木霉表达纤维素酶
2016-09-10 06:54:52薛鲜丽罗会颖张永杰苏小运
食品工业科技 2016年11期
王 榕,宫 莉,姚 斌,薛鲜丽,罗会颖,张永杰,苏小运,*
(1.山西大学生命科学学院,山西太原 030006;2.中国农业科学院饲料研究所,北京 100081)
利用RNAi抑制cre1基因转录提高里氏木霉表达纤维素酶
王榕1,2,宫莉2,姚斌2,薛鲜丽2,罗会颖2,张永杰1,*,苏小运2,*
(1.山西大学生命科学学院,山西太原 030006;2.中国农业科学院饲料研究所,北京 100081)
本研究采用RNA干扰技术(RNAi)对里氏木霉主要的转录抑制因子cre1基因的表达进行抑制,以期提高里氏木霉生产纤维素酶的能力。通过PCR,从里氏木霉的基因组中扩增得到cbh1启动子、反向的cre1基因片段(568~963 bp)、正向的cre1基因片段(655~961 bp)和cbh2终止子,利用DNA assembler方法将这些基因连接到pRS424质粒上,构建成pCre1-i质粒。再将RNAi盒分作两个片段扩增,同时转化里氏木霉并通过PCR鉴定阳性转化子。摇瓶发酵显示其中一株转化子在纤维素诱导培养基中培养4.5 d后,其纤维素酶的滤纸酶活、内切葡聚糖酶活和CBHI酶活为0.67、3.70和0.46 U/mL,较出发菌株分别提高了1.3、1.8和5.6倍。通过实时荧光RT-PCR检测,发现该转化子中cre1基因的转录水平比出发菌株降低了43%。
cre1,RNAi,纤维素酶,DNA assembler
纤维素酶作为自然界中数量最大的可再生资源,在制酒、酱油酿造、饮料加工等食品行业有广泛的应用价值[1]。目前,报道的主要产纤维素酶的菌株多为真菌[2]。其中,里氏木霉因其表达能力强(工业上,经改造后的里氏木霉,其产纤维素酶的产量可达到100 g/L以上[3])、易于培养和符合“一般公认安全”等优点,在工业表达酶基因、尤其是纤维素酶上得到了极大的应用。里氏木霉的纤维素酶系包括两个外切纤维素酶、五个内切纤维素酶、一个β-葡萄糖苷酶和六个裂解性多糖单加氧酶[4-5]。
cre1是里氏木霉的一个广域的转录抑制因子,能调节大部分纤维素酶甚至半纤维素酶基因的表达[6]。在里氏木霉主要的纤维素酶和木聚糖酶基因上,都或多或少的存在着cre1转录因子的结合位点[7]。虽然cre1的主要作用是转录抑制因子,但完全敲除cre1基因,在以微晶纤维素做唯一碳源进行诱导时,产酶水平低于野生型菌株的最大产酶水平,意味着cre1还具有一定的转录激活作用[8]。因此,对cre1基因的调控,不能简单的采取基因敲除的策略,而应该采取抑制其表达的策略。RNA干扰(RNAi)技术提供了一种快速、有效抑制目的基因表达的方法[9]。受到RNAi的受体菌株,其被调控基因呈现从高到低的一系列表达水平,与基因敲除在性状上呈现有或无不同,其具有连续性。
本研究采取RNAi技术对cre1基因进行干扰调节,期望能提高受体菌株产纤维素酶的水平,为利用RNAi技术对里氏木霉进行遗传改造构建高效表达的菌株提供新的思路,同时为纤维素酶在食品领域的广泛应用提供可能性。
1 材料与方法
1.1材料与仪器
里氏木霉QM9414(ATCC 26921)和Tu6ΔtKU70(ATCC MYA-256,尿嘧啶缺陷菌株)[10]均由中国科学院微生物研究所董志扬教授馈赠。pEASY-T3质粒购于全式金;质粒pRS424为本实验室保存,用作质粒骨架构建。pCre1-i重组质粒(pRS424-armL-cbh1p-cre1f-cre1r-cbh1t-pyrG-armR,图1)由本实验室构建。
里氏木霉生长培养基:(NH4)2SO45.0 g/L,KH2PO415.0 g/L,MgSO4·7H2O 0.6 g/L,CaCl2·2H2O 0.6 g/L,CoCl2·6H2O 0.0037 g/L,FeSO4·7H2O 0.005 g/L,ZnSO4·7H2O 0.0014 g/L,MnSO4·H2O 0.0016 g/L,葡萄糖20 g/L,自然pH。
里氏木霉纤维素诱导培养基:(NH4)2SO45.0 g/L,KH2PO415.0 g/L,MgSO4·7H2O 0.6 g/L,CaCl2·2H2O 0.6 g/L,CoCl2·6H2O 0.0037 g/L,FeSO4·7H2O 0.005 g/L,ZnSO4·7H2O 0.0014 g/L,MnSO4·H2O 0.0016 g/L,微晶纤维素Avicel(购自Sigma公司)20 g/L,自然pH。
色氨酸(Trp)缺陷型培养基:YNB 6.7 g/L,葡萄糖 20 g/L,Trp Do supplement(购自Clontech)0.64 g/L,琼脂粉 20 g/L。
T100TM普通PCR仪、CFX-96实时荧光定量PCR仪和212PRO凝胶成像仪购自美国Bio-Rad公司;CR22GШ立式高速冷冻离心机购自日本HITACHI公司;移液枪购自德国Eppendorf公司;CT6E台式高速冷冻离心机购自日本HITACHI公司;SPECORD200PLUS紫外分光光度计购自德国耶拿分析仪器公司;H1MFDG酶标仪购自美国BioTek仪器有限公司;SPX-250生化培养箱购自北京利康达圣科技术发展有限公司;ZHWY-211C恒温培养振荡器购自上海智诚分析仪器制造有限公司。
1.2实验方法
1.2.1pCre1-i质粒的构建以里氏木霉QM9414基因组为模板,利用引物ParmL(SR)F1/R1、PpyrG(SR)F1/R1、ParmR(SR)F1/R1(表1)扩增获得armL、pyrG(乳清酸核苷-5′-磷酸脱羧酶)和armR。armL和armR片段分别作为在里氏木霉里进行同源重组的左臂和右臂,pyrG基因作为尿嘧啶营养缺陷型的筛选标记基因。并同时引进NotI和EcoR I酶切位点。利用引物Pcbh1P(SR)F1/R1、Pcre1(SR)F1/R1、Pcre1(SR)F2/R2、Pcbh2t(SR)F1/R1,扩增获得cbh1启动子、反向的cre1基因(cre1r)568~963 bp片段、正向的cre1基因(cre1f)655~961bp片段以及cbh2终止子并同时引入KpnI和EcoR I酶切位点。将pRS424质粒用EcoR I酶切,电泳、回收线性化质粒载体。将其与armL、pyrG、armR按1∶3∶3∶3的摩尔比用氯化锂法共同转化酿酒酵母AH109的感受态细胞,采用DNA assembler方法进行拼接[11]。DNA assembler利用酿酒酵母的高效同源重组机制,能快速的将多个具有相同核苷酸末端(重叠区长度需要大于30 bp)的DNA片段在体内一次性和线性化的质粒拼接起来。同样的方法将EcoRI酶切的pRS424质粒与cbh1启动子、反向的cre1基因、cbh2终止子DNA片段,共转化酿酒酵母AH109的感受态细胞,涂布在色氨酸(Trp)缺陷型培养基上,30 ℃静置培养48 h。对酵母菌落做菌落PCR,以鉴定是否已经成功构建pRS424-armL-pyrG-armR和pRS424-cbh1p-cre1f-cbh2t重组质粒。将PCR鉴定为阳性的酵母菌落挑取并接种于3 mL YPD培养基上,30 ℃、180 r/min振摇培养过夜,提取质粒。将重组质粒转化大肠杆菌Trans1感受态细胞(全式金,北京),涂布于含100 μ g/mL氨苄的LB琼脂平板上,37 ℃静置培养16 h。挑取单克隆菌落,接种于3 mL LB培养基(含100 μg/mL氨苄)中,37 ℃、180 r/min振摇培养过夜,提取质粒。
随后,用KpnI和EcoRI双酶切pRS424-cbh1p-cre1r-cbh2t,同时将用PCR扩增所得到的正向的cre1基因用KpnI和EcoR I双酶切,将回收质粒和正向的cre1基因用T4连接酶(TaKaRa公司)连接、转化大肠杆菌Trans1感受态细胞,筛选重组质粒pRS424-cbh1p-cre1r-cre1f-cbh2t(图1)。利用引物Pcbh1P(SR)F3/Pcbh2t(SR)R3从重组质粒pRS424-cbh1p-cre1反-cre1正-cbh2t上扩增获得cbh1p-cre1r-cre1f-cbh2t片段。
最后,将pRS424-armL-pyrG-armR重组质粒用EcoR I酶切,与上一步获得的cbh1p-cre1r-cre1f-cbh2t片段用DNA assembler法进行重组质粒的构建。经验证正确后,最终将重组质粒转化大肠杆菌Trans1感受态细胞,提取重组质粒pCre1-i(图1)。

表1 本研究所用引物
1.2.2里氏木霉的转化用ParmLF2/PpyrGR2和Pcbh2tF2/ParmRR2(表1)引物对分别从重组质粒pCre1-i上扩增armL-cbh1p-cre1r-cre1f-cbh2t-pyrG和cbh2t-pyrG-armR片段。在1%琼脂糖胶上电泳,切下PCR产物中的目的条带,纯化DNA备用。
将里氏木霉TU6ΔtKU70接种于土豆培养基(PDA)平板上,30 ℃静置培养7 d待其产孢,将孢子刮下并接种于100 mL MM-glucose培养基中,30 ℃、180 r/min振摇培养过夜。在12层纱布上过滤收集萌发的菌丝,加入10 mg/mL的Lysing enzymes(购自Sigma公司),在30 ℃消化1~2 h。收集原生质体,将armL-cbh1p-cre1r-cre1f-cbh2t-pyrG和cbh2t-pyrG-armR片段混合,以PEG介导的原生质体转化法将两种DNA同时转入里氏木霉TU6ΔtKU70菌株。每微克DNA可获得约10个转化子,转化子中同时含有两种DNA的转化子的转化效率为80%。
1.2.3重组转化子的筛选转化子在含1 mol/L山梨醇的MM-glucose琼脂培养基上生长和选择。7 d后,挑取单个克隆,接种于MM-glucose培养基上进行再次生长、选择。挑取单克隆接种于PDA平板上,30 ℃培养5~6 d。取部分菌丝,用UniversAll-tissue extraction PCR kit(YEASTERN,Taipei,Taiwan)提取基因组DNA,通过PCR验证cre1基因是否转入以及两片段是否发生重组并成功整合到里氏木霉基因组。PCR所用引物为Pcbh1P(SR)F1/YZ-pyrGR(表1)。
1.2.4转化子的诱导培养将出发菌株TU6ΔtKU70的孢子和各转化子的孢子,接种2.0 mL 1×107/mL的孢子于100 mL MM-glucose培养基中。30 ℃、180 r/min振摇培养1 d。将菌丝用12层纱布过滤,收集菌丝并用大量无菌水冲洗,以去除残余的葡萄糖。
称取等量(900 mg)的菌丝,接种于100 mL MM-Avicel液体培养基中,出发菌株和每个转化子均做3个平行。30 ℃、180 r/min振摇培养7 d以诱导纤维素酶的生产。从第3 d开始,每12 h收集发酵液,储存于4 ℃冰箱备用。
1.2.5纤维素酶活测定纤维素酶滤纸酶活的测定:纤维素酶的滤纸酶活(FPase activity)采用Whatman一号滤纸,参照IUPAC标准方法进行测定。滤纸酶活的单位定义为:1 mL液体酶,在50 ℃、pH4.8的条件下,每小时水解滤纸底物,产生1 mg还原糖(以葡萄糖计)所需要的酶量定义为一个酶活力单位(U)。
外切纤维素酶活的测定:用4-methylumbelliferyl-β-D-cellobioside(MUC,Sigma)作底物,称取45 mg MUC,溶于3 mL DMSO(Sigma),再将其转移到42 mL柠檬酸缓冲液(pH5.2,20 mmol/L)中。将25 μL适当稀释的酶液和250 μL MUC、25 μL葡萄糖(1 mol/L)混合,此为不加纤维二糖实验组。在混合溶液中,葡萄糖会抑制BG降解MUC,因此,测得的活性实际为CBH1和EG的活性。同时设加纤维二糖实验组:25 μL酶液和200 μL MUC、25μL(50 mmol/L)纤维二糖和葡萄糖于50 ℃反应10 min。加入纤维二糖会抑制CBH的活性,因此测得的是EG的活性,于50 ℃反应10 min。加入250μL Na2CO3(1 mmol/L),于370 nm测定吸光度。将不加纤维二糖实验组OD值减去加纤维二糖实验组,即为外切纤维素酶较为特异的活性。一单位的外切纤维素酶酶活定义为每秒催化1 nmol/L MUC水解所需要的酶量。
内切纤维素酶酶活测定:采用1%羧甲基纤维素钠(CMC)作为底物进行测定。取酶液62.5 μL,加入到175 μL磷酸—柠檬酸钠缓冲液(0.05 mol/L、pH4.8)配制的CMC中,加水定容到250 μL。50 ℃,反应1 h,测定酶活力。CMC酶活的单位定义为:1 mL液体酶,在50 ℃、pH4.8的条件下,每小时水解羧甲基纤维素钠,产生1 mg还原糖(以葡萄糖计)所需要的酶量定义为一个酶活力单位(U)。
β-葡萄糖苷酶酶活的测定:用pNP-Glucose(pNPG)为底物,取0.24 g pNPG用蒸馏水定容到50 mL,得到16 mmol/L的pNPG为底物。使用前-20 ℃避光保存。取50 μL已稀释的酶液,加入15.625 μL pNPG和125.5 μL柠檬酸-磷酸氢二钠缓冲液(pH5.0)用水定容到250 μL,pNPG终浓度为1 mmol/L,振荡混匀。50 ℃水浴保温1 h,迅速冷却向各试管中加入750 μL Na2CO3试剂,再向空白中补加50 μL酶液,混合均匀。以0号管为参比,测定540 nm的吸光度。β-葡萄糖苷酶酶活的单位定义为:1 mL液体酶,在50 ℃、pH5.0的条件下,每小时水解pNPG,产生1 mg还原糖(以葡萄糖计)所需要的酶量定义为一个酶活力单位(U)。
对出发菌株TU6ΔtKU70和转化子,经预实验测定酶活,发现在4.5 d时纤维素酶的酶活最高。因此,选取4.5 d的发酵液,分别测定总体纤维素酶酶活(滤纸酶酶活)及各分解酶酶活包括内切纤维素酶酶活(CMC酶活)、外切纤维素酶CBHI的酶活以及β-葡萄糖苷酶酶活(BG酶活)。
1.2.6cre1的转录丰度测定出发菌株与重组菌株先接种2.0 mL 1×107/mL的孢子于100 mL MM-glucose培养基中。30 ℃、180 r/min振摇培养1 d。称取等量湿重(900 mg)的菌丝,接种于100 mL MM-纤维素液体培养基中,30 ℃、180 r/min振摇培养2 d以诱导纤维素酶的生产。收集诱导后第2 d的菌丝,在滤纸上压干菌丝,进行研磨并提取总RNA。将所提取总RNA用ReverTra Ace-α-试剂盒(TOYOBO,日本)处理,将其中的mRNA反转录为cDNA。设计引物,以actin 基因作为内参,q-cre1F/q-cre1R为引物(表1),1 μg cDNA为模板,按TransStart Tip Green qPCR SuperMix(全式金.北京)试剂盒进行操作,比较转化子和对照株中cre1转录物的相对丰度。
1.3数据处理方法
所有结果均釆用Excel和SPSS16.0版统计软件中One-Way ANOVA过程进行方差及其显著性分析,p<0.05表示差异显著,p<0.01表示差异极显著。
2 结果与分析
2.1pCre1-i质粒构建及重组子的鉴定
按1.2.1的方法,构建了pCre1-i质粒(图1),并通过Not I酶切验证重组质粒是否构建成功(图3A)。通过ParmLF2/PpyrGR2和Pcbh2tF2/ParmR2(表1)引物对从重组质粒pCre1-i扩增获armL-cbh1p-cre1r-cre1f-cbh2t-pyrG和cbh1t-pyrG-armR片段,将两个片段共转化里氏木霉TU6ΔtKU70(图2)。所采用的宿主菌为ku70基因敲除株,并且用于转化的两个片段间设计了1 kb的重叠片段,因此,预期两个片段可以重组在一起,并在左、右重组臂(armL和armR)的介导下,整合到里氏木霉基因组的ku70座位上。cbh1启动子驱动含反、正cre1基因片段的转录,mRNA在细胞内形成发夹状RNA并在Dicer的作用下形成小分子RNA并最终使降解目的基因cre1的mRNA降解(图2)。设计一对引物Pcbh1P(SR)F1和YZ-pyrGR(表1),PCR验证是否armL-cbh1p-cre1反-cre1正-cbh2t-pyrG和cbh2t-pyrG-armR片段已成功整合(图3B)。从图3B可以看出,六个转化子均有3 kb左右的特异扩增条带,与预期条带大小一致,而以出发菌株做模板则没有此条带,说明两个DNA片段已经成功转入里氏木霉TU6ΔtKU70并整合在一起。

图1 pCre1-i质粒构建示意图Fig.1 The diagram for pCre1-i plasmid construction
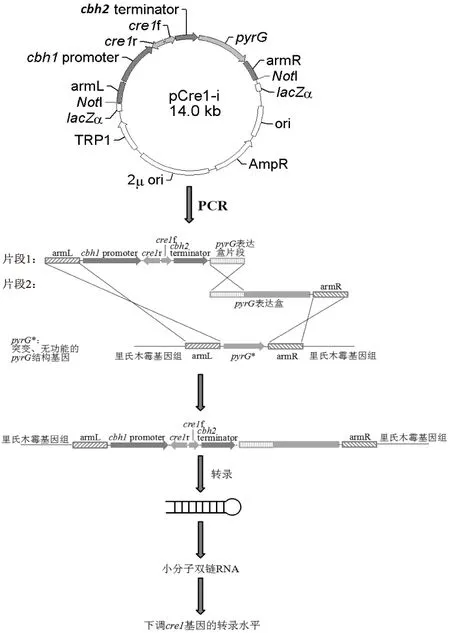
图2 通过RNAi抑制里氏木霉cre1的表达示意图Fig.2 The schematic diagram of inhibiting cre1 expression by RNAi

图3 PCR鉴定目的片段是否插入里氏木霉基因组Fig.3 Identification of positive Trichoderma reesei transformantswith the target fragment by PCR注:A:pCre1-i质粒酶切验证。M:DNA分子量标准;1:未酶切pCre1-i质粒;2:Not I酶切后的pCre1-i质粒。B:PCR验证RNAi片段重组入里氏木霉基因组。M:DNA分子量标准;1:出发菌株TU6ΔtKU70;2-7:各转化子。
2.2纤维素酶活测定
经预实验,从6个转化子中选择一株酶活最高的2号转化子进行详细的纤维素酶酶活测定。分别以滤纸、CMC、MUC和pNPG为底物测定出发菌株和2号转化子的总体纤维素酶酶活、内切纤维素酶酶活、外切纤维素酶CBHI酶活以及β-葡萄糖苷酶的酶活。从图4可看出,2号转化子的总体纤维素酶活是TU6ΔtKU70的1.3倍,说明转化子较出发菌株,其纤维素酶酶活有显著提高。与出发菌株相比,该转化子的内切纤维素酶活性提高极为显著,为出发菌株的1.8倍(图4)。外切纤维素酶CBHI活性也有一定程度的提高,2号转化子的CBHI酶活是出发菌株的5.6倍(图4)。但对β-葡萄糖苷酶而言,转化子的酶活与TU6ΔtKU70相比下降了65%(图4)。

图4 转化子2与出发菌株TU6ΔtKU70酶活比较Fig.4 Comparison of enzyme activities of Transformant No. 2 with those of the parent strain TU6ΔtKU70
2.3cre1的表达差异研究
将出发菌株和转化子取等量湿重菌丝,用MM-纤维素培养基30 ℃诱导表达。提取48 h的RNA并反转获得cDNA,通过荧光定量PCR(q-PCR),以actin基因为内参,对cre1转录水平进行监测。荧光定量 PCR 结束后,通过 IQ5 软件自动分析得出目的基因和内参基因的Ct值,采用2-ΔΔCT方法,比较转化子和对照株中cre1基因的相对转录丰度。结果显示,与TU6ΔtKU70相比,转化子cre1表达量下降了43%(图5)。这说明正如预期的一样,cre1的表达得到了一定程度的抑制,从而提高了里氏木霉生产纤维素酶的能力。

图5 出发菌株TU6ΔtKU70与转化子2中cre1的转录丰度比较Fig.5 Comparison of the transcript abundance of cre1 between the parent strain TU6ΔtKU70and Transformant No. 2注:A:cre1和看家基因actin的扩增曲线。B:cre1的转录丰度比较。
3 结论与讨论
据报道,目前已经有30多种丝状真菌中可以应用RNA干扰技术[12],这种技术对于某些重要的管家基因的功能研究来讲是一种很理想的工具,在里氏木霉里也曾经得到过成功应用。例如,将cbh1基因的表达进行RNAi的抑制,可以提高外源基因黑曲霉脂酶在里氏木霉里的分泌表达[13]。通过RNAi,成功将cre1基因的表达进行下调,证明RNAi可用于对转录因子的调控。相应的,与出发菌株相比,2号转化子的总体纤维素、内切纤维素酶和外切纤维素酶的酶活均有明显的提高,将有利于纤维素酶在食品工业更为广泛和深入的应用。其中,CBHI的活性提高了5.6倍。有意思的是,β-葡萄糖苷酶的活性下降了。里氏木霉的β-葡萄糖苷酶活性来自于bgl1基因,和cbh1启动子有4个功能性的cre1结合位点不同,虽然bgl1启动子上也有1个可能的cre1结合位点,但看起来bgl1的表达并不受cre1基因的调节抑制,这也许可以解释在2号转化子中,β-葡萄糖苷酶的活性的原因。
在构建RNAi载体时,我们选用了DNA assembler的方法[11],在酿酒酵母AH109中,进行体内重组以将各目的片段拼接起来。与常规依赖于限制性酶切和连接酶连接不同,它是利用同源重组的方法使具有同源末端的片段连接起来,能够实现同时对多个片段的连接,大大提高了构建复杂、含多个基因片段载体的效率。另外,我们在构建好质粒载体pCre1-i后,通过PCR将cre1基因的RNAi盒以及两侧的同源重组臂分作两个片段PCR扩增出来,并同时转化里氏木霉,获得了在里氏木霉细胞内两个片段重组在一起的菌株。因此,这还为以后将多个基因片段(如合成生物学研究中的代谢途径中的多个基因)转化入里氏木霉里并在其中进行同源重组打下了良好的基础。
[1]赵国萍,李迎秋.纤维素酶的研究进展及其在食品工业的应用[J].山东食品发酵,2015,(2):37-40.
[2]张加春,王权飞,余尊祥.里氏木霉的纤维素酶产生条件研究[J].食品与发醉工业,2003,(03):21-23.
[3]Su X,Schmitz G,Zhang M,et al. Heterologous gene expression in filamentous fungi[J]. Advances in Applied Microbiology,2012,81(1):1-61.
[4]Martinez D,Berka RM,Henrissat B,et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei(syn. Hypocrea jecorina)[J]. Nature Biotechnology,2008,26(5):553-560.
[5]Hakkinen M,Arvas M,Oja M,et al. Re-annotation of the CAZy genes of Trichoderma reesei and transcription in the presence of lignocellulosic substrates[J]. Microbial Cell Factories,2012,11:134.
[6]Strauss J,Mach RL,Zeilinger S,et al. The carbon catabolite repressor protein from Trichoderma reesei[J]. FEBS Letters,
1995,376(1-2):103-107.
[7]苏小运.嵌合体转录激活因子增强里氏木霉的纤维素酶、半纤维素酶和其它基因转录的研究.[D]北京:中国科学院博士论文,2009.
[8]Portnoy T,Margeot A,Seidl-Seiboth V,et al. Differential regulation of the cellulase transcription factors XYR1,ACE2,and ACE1 in Trichoderma reesei strains producing high and low levels of cellulase[J].Eukaryotic Cell,2011,10(2):262-271.
[9]Su X,Qin L,and Dong Z. Chapter 15-RNAi-Mediated Gene Silencing in Trichoderma:Principles and Applications//Biotechnology and Biology of Trichoderma[M]. Waltham,USA:Elsevier,2014:215-226.
[10]Guangtao Z,Hartl L,Schuster A,et al. Gene targeting in a nonhomologous end joining deficient Hypocrea jecorina[J]. Journal of Biotechnology,2009,139(2):146-151.
[11]Shao Z,Zhao H. DNA assembler,aninvivogenetic method for rapid construction of biochemical pathways[J]. Nucleic Acids Research,2009,37(2):e16.
[12]Kuck U,Hoff B. New tools for the genetic manipulation of filamentous fungi[J]. Applied Microbiology and Biotechnology,2010,86(1):51-62.
[13]Qin L,Cai F,Dong X,et al. Improved production of heterologous lipase in Trichoderma reesei by RNAi mediated gene silencing of an endogenic highly expressed gene[J]. Bioresource Technology,2012,109:116-122.
Improving cellulase expression ofTrichodermareeseiby RNAi-mediated repression ofcre1 transcription
WANG Rong1,2,GONG Li2,YAO Bin2,XUE Xian-li2,LUO Hui-ying2,ZHANG Yong-jie1,*,SU Xiao-yun2,*
(1.School of Life Sciences,Shanxi University,Taiyuan 03006,China;2.Key Laboratory for Feed Biotechnology of the Ministry of Agriculture,Feed Research Institute,Chinese Academy of Agricultural Sciences,Beijing 100081,China)
The RNA interference(RNAi)technique was used to repress the expression of the main transcriptional repressorcre1 in order to improve the cellulase production ofTrichodermareesei. Thecbh1 promoter,a reversedcre1 gene fragment(568~963 bp),an ordinarycre1 gene fragment(655~961 bp),andcbh2 terminator were all amplified from the genomic DNA ofT.reeseiby polymerase chain reaction(PCR). The DNA assembler method was used to assemble all these gene fragments in pRS424 to obtain the pCre1-i plasmid. Two fragments encompassing the RNAi cassette were amplified from the plasmid and transformed simultaneously intoT.reesei. PCR was used to verify the positive colonies. In the flask batch culture,one of the transformants displayed 0.67、3.70 and 0.46 U/mL for the filter paper cellulase,endoglucanase,and CBHI activity,which were 1.3,1.8,and 5.6 folds of the parent strain. By using RT-qPCR,the transcript level ofcre1 in this transformant was determined to be lowered to 43% of that of the parent strain.
cre1;RNAi;cellulase;DNA assembler
2015-12-02
王榕(1990-),女,硕士研究生,研究方向:食品科学,E-mail:wangrong109@163.com。
苏小运(1979-),男,博士,研究员,研究方向:丝状真菌基因工程,E-mail:suxiaoyun@caas.cn。
张永杰(1979-),男,博士,教授,研究方向:微生物进化生物学与分子生态学,E-mail:zhangyj2008@sxu.edu.cn。
国家自然科学基金青年基金项目(31400067);中国农业科学院青年英才计划。
TS201.3
A
1002-0306(2016)11-0189-06
10.13386/j.issn1002-0306.2016.11.031
猜你喜欢
中国特种设备安全(2021年7期)2022-01-19 05:07:32
湖南农业大学学报(自然科学版)(2021年3期)2021-07-02 01:34:24
食品与机械(2019年1期)2019-03-30 01:14:40
中国科技信息(2016年19期)2016-10-25 08:15:14
应用化工(2014年12期)2014-08-16 13:10:46
中国酿造(2014年9期)2014-03-11 20:21:06
汽车零部件(2014年2期)2014-03-11 17:46:34
湖南农业科学(2014年5期)2014-02-27 14:29:42
大学化学(2013年2期)2013-02-13 09:25:54
